La tétracycline, connue sous le nom commercial Sumycin, agit en bloquant la fixation de l’ARNt sur la sous-unité 30S ribosomale, interrompant l’élongation de la chaîne protéique bactérienne. Ce mécanisme confère une activité sur un spectre large, incluant bactéries Gram positives, Gram négatives, rickettsies et spirochètes. Sa biodisponibilité digestive varie selon la prise alimentaire et les interactions avec les ions divalents comme calcium et magnésium. Sa diffusion tissulaire est importante, notamment dans les voies respiratoires et génito-urinaires. L’élimination se fait par voie rénale et biliaire. Les effets indésirables incluent photosensibilisation, troubles digestifs et coloration dentaire en cas d’administration précoce. Les guides thérapeutiques mentionnent sumycin prix, en soulignant la nécessité de restreindre son utilisation afin de limiter les résistances acquises.
Biologia.uniroma1.it
European Journal of Clinical Investigation (2005) 35, 82– 92 Peroxisome proliferator-activated receptor γ: the more the merrier? C. A. Argmann*, T.-A. Cock* and J. Auwerx*† *Institut de Génétique et de Biologie Moléculaire et Cellulaire, CNRS/INSERM/Université Louis Pasteur, 67404 Illkirch,
France; †Institut Clinique de la Souris, Génopole Strasbourg, 67404 Illkirch, France
Abstract
The consequence of activating the nuclear hormone receptor, peroxisome proliferator-activated receptor gamma (PPARγ), which coordinates adipocyte differentiation, validatesthe concept, ‘you are what you eat’. Excessive caloric intake leads to fat formation if theenergy from these nutrients is not expended. However, this evolutionary adaptation to storeenergy in fat, which can be released under the form of fatty acids, potent PPARγ agonists,has become a disadvantage in today’s affluent society as it results in numerous metabolicimbalances, collectively known as the metabolic syndrome. With the surge of human andgenetic studies on PPARγ function, the limitations to the benefits of PPARγ signalling havebeen realized. It is now evident that the most effective strategy for resetting the balance ofthis thrifty gene is through its modulation rather than full activation, with the goal to improveglucose homeostasis while preventing adipogenesis. Finally, as more PPARγ targetedpathways are revealed such as bone homeostasis, atherosclerosis and longevity, it is mostcertain that the PPARγ thrifty gene hypothesis will evolve to incorporate these. Keywords Atherosclerosis, longevity and bone, metabolism, mouse models, PPARγ. Eur J Clin Invest 2005; 35 (2): 82– 92 PPARγ in a westernised society
and dietary pressures for which no adaptation has beenpossible in such a short time [1]. Besides the increase in
Coinciding with the modernisation of society was the emerg-
availability and intake of calories, it is predicted that a number
ence of the western lifestyle diseases including obesity and
of crucial nutritional characteristics of our ancestral diet
the metabolic diseases that are associated with it such as
have been fundamentally altered during the Neolithic and
hyperlipidemia; insulin resistance; type 2 diabetes mellitus
industrial era including: the glycaemic load; fatty acid
(T2DM) and cardiovascular disease. Because genetically,
balance; macronutrient balance; trace nutrient density;
our bodies are viewed as being virtually identical to what
acid–base balance; sodium–potassium balance and fiber
they were some 20 000 years ago, it is believed that the
content [1]. Because thrifty metabolism was evolutionarily
appearance of agriculture, the domestication of animals and
programmed to coordinate cycles of feast or famine and
the industrial revolution have created new environmental
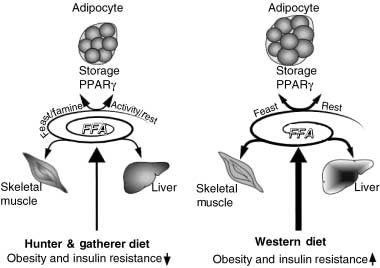
physical activity or rest (Fig. 1), discordance has now beencreated between our lifestyle, and the genes, which aresuited to them [1,2]. One gene that has been identified at
Institut de Génétique et de Biologie Moléculaire et Cellulaire,
the centre of this feed forward pathway that favours energy
CNRS / INSERM / Université Louis Pasteur, 67404 Illkirch, France
storage by adipocytes is PPARγ. As PPARγ activity is governed
( C.-A. Argmann, T.-A. Cock, J. Auwerx); Institut Clinique de la
by the binding of small lipophilic ligands, mainly fatty acids
Souris, Génopole Strasbourg, 67404 Illkirch, France ( J. Auwerx)
derived from nutrition or meta-bolism (reviewed in [2] and
Correspondence to: Johan Auwerx, The Institut de Génétique et
[3]), it is not unlikely that the level of PPARγ activity has
de Biologie Moléculaire et Cellulaire, 1 rue Laurent Fries, F-67404
been altered throughout evolution. Because the activation
Illkirch, France. Tel.: +33 388653425; fax: +33 388653201,
of PPARγ leads to adipocyte differentiation and fatty acid
storage, the exposure of people to prolonged chronic levels of
Received 11 November 2004; accepted 30 November 2004
fatty acid-like PPARγ ligands, akin to the westernised lifestyle,
PPARγ, the more the merrier? 83 Human genetic variants
PPARγ is mainly known for its role in adipogenesis, anobservation based on molecular and cellular studies thatshowed that the expression of PPARγ in cells is sufficient toinduce adipocyte differentiation [9]. Moreover, targetedmutagenesis of the PPARγ gene in embryonic stem (ES) cellsand knocking down the endogenous PPARγ2 in cell linesconfirmed the commanding role of PPARγ in adipocyte dif-ferentiation [10,11]. Consistent with this, PPARγ has beenshown to increase the expression of genes that promote fattyacid storage, whereas it repressed genes that induce lipolysisand the release of free fatty acids (FFAs) in adipocytes [2,12].
PPARγ’s role in adipogenesis in man has been underscored
by human genetic association studies that tie the PPARGlocus on chromosome 3p25-p24 with obesity in Pima Indi-
Figure 1 The impact of dietary input and lifestyle on peroxisome
ans [13]. More striking is that of the more than 40 different
proliferator activated receptor gamma (PPARγ) activity and the
reported associations of genetic variation and population
balancing act of lipids and tissues in energy homeostasis. Our biochemical cycle has been evolutionarily programmed to
risk to T2DM, the most widely reproduced association is
coordinate cycles of feast with famine and physical exercise. One
that of the Pro12Ala polymorphism in PPARγ [14,15]. The
candidate gene that has been identified at the center of this feed
less common alanine allele was originally reported to induce
forward pathway that favours energy storage by adipocytes is
an impressive 70% reduction in risk in T2DM in Finnish
PPARγ. The interactions between the main metabolic tissues that
and second-generation Japanese populations [16], a finding
store (adipose tissue) and oxidize (skeletal muscle and liver) free
subsequently confirmed in a large independent study [17].
fatty acids (FFAs) are influenced by dietary-derived FFAs and
Although this association was initially challenged by four
adipokines. Adipokines are subdivided into insulin sensitising (e.g.
subsequent ‘negative studies’, a recent metaanalysis of all
adiponectin) and insulin resistance (tumour necrosis factor α)
published data involving over 25 000 cases of diabetes, has
adipokines. The thickness of the arrows reflects the force of the
unequivocally confirmed these data and suggests that
effects. Two different conditions are compared, the response to the hunter and gatherer diet and to the western lifestyle.
patients who carry the proline allele have an odds ratio of1·27 of developing T2DM [14]. The risk allele has only amodest effect on an individual basis; however, the proline
through a feed-forward pathway would result in obesity [4 –
allele is very common, especially in European populations
(75%) and in terms of a population-attributable risk, it isa staggering 25% [14]. The broad impact of this variant onthe risk of T2DM and its unique localization to the NH2terminus (responsible for ligand-independent transcrip-
Thrifty metabolism
tional activity) (Fig. 2) makes understanding how thisvariant affects insulin sensitivity important for accelerating
Obesity is more prevalent in affluent societies and so too
the development of novel pharmacological agents. At the
are the metabolic diseases, such as the metabolic syndrome,
molecular level, the Pro12Ala polymorphism, which introduces
which put a heavy social and economic burden on society.
a missense change in the coding region of the PPARγ2 gene,
Strategies to lessen the disease burden include diet and exercise
in vitro, is suggested to induce a partial loss of function as
regimes as well as the rigorous treatment of hypertension,
a result of decreased DNA-binding affinity and reduced
dyslipidemia and hyperglycaemia. Ironically, the synthetic
transcriptional activity [16,18](Fig. 2). Because genes do
PPARγ ligands such as thiazolidinediones (TZDs), which
not work in a vacuum, the potential for gene–environment
increase the body’s sensitivity to insulin, have the unfortunate
interaction must also be considered, especially as some find-
side effect to also promote fat accretion [7,8]. The long-term
ings for the Pro12Ala PPARγ variant have been reported to
consequences of this are unknown. Moreover, the mecha-
differ depending on the superimposition of environmental
nism by which TZDs act and why they are effective is still
factors, such as obesity and the ratio of unsaturated to sat-
not understood. In this review we summarize the studies
urated fatty acids [19,20]. The Pro12Ala PPARγ variant has
that shed new light on the role of PPARγ in adipose tissue
also been associated with other phenotypes such as longevity,
homeostasis and emphasize that insulin sensitization can be
hypertension and birth weight, which may provide further
achieved without the concomitant increase in fat deposition by
mechanistic insights into the mechanisms of this variant
modulating PPARγ activity. In addition to obesity however,
in vivo [21–23]. In addition to the Pro12Ala PPARγ variant,
altered PPARγ activity through our westernised lifestyle has
even more compelling evidence for a link between insulin
potentially influenced bone homeostasis, atherosclerosis
sensitivity and PPARγ in humans has come from studying
risk and longevity, as recent literature supports their signif-
individuals with dominant negative/complete loss of func-
icant regulation by PPARγ. Thus, in this review we have also
tion mutations occurring in the ligand-binding domain. The
highlighted the role of PPARγ in nonadipose tissue.
dominant negative/loss of function mutations that have been
2005 Blackwell Publishing Ltd, European Journal of Clinical Investigation, 35, 82– 92
together with the characterization of mice chimaeric forPPARγ–/– ES cells [11], showed the importance of PPARγin adipose tissue development in vivo. Until the past year,determination of the physiological function of PPARγin mice had been limited to studies in the heterozygousPPARγ+/– mice, which was a difficult model to comprehendas these mice are resistant to the obesity and insulin resist-ance that is induced by a high-fat diet [31,32]. To overcomethe embryonic lethality, mice with tissue-specific deletionsof PPARγ have been generated to help elucidate the tissuespecific activities of PPARγ (summarized in Table 1).
The specific reduction of PPARγ1 and PPARγ2 in the adiposetissue revealed the essential role of PPARγ in adipogenesis[33,34]. Moreover, the essential role of adipose tissue inwhole body metabolism was exemplified by the significant
Figure 2 Genetic and pharmacological evidence supporting
mortality rate (> 40%) of the WAT-specific hypomorphic
PPARγ’s relationship with adipogenesis and insulin sensitivity.
PPARγ1 and PPARγ2 knockdown mice (PPARγ hyp/hyp),
Human mutations, mouse models and pharmacological studies
which were severely lipodystrophic [33]. When an aP2-driven
demonstrate that the level of PPARγ activity directly corresponds
Cre recombinase transgene was used to excise PPARγ1 and
to adipose mass (adipogenesis). By contrast, insulin sensitivity can
PPARγ2 from the mature adipocytes, a more moderate
be achieved by both inhibition and activation of PPARγ, as
reduction of adipose mass was observed, which was accom-
illustrated by the human mutations and pharmacological studies.
panied by hyperlipidemia and liver steatosis [34]. This was
Pro495Leu mutation is equivalent to Pro467Leu mutation in human PPARγ1.
in contrast to the surviving adult PPARγhyp/hyp mice, whichdid not have liver steatosis or dyslipidemia [33]. Liver stea-tosis or dyslipidemia were predominantly prevented in thePPARγhyp/hyp mice by efficient oxidation of excess lipids in
identified include Phe388Leu, Pro467Leu (also known as
the muscle by PPARα- and PPARβ/δ-driven pathways.
Pro495Leu in PPARγ2), and Arg425Cys, which have been
Intriguingly, both adipose PPARγ-deficient models had
associated with partial lipodystrophy resulting in loss of
relatively normal glucose tolerance [33,34]. Confirmation
fat from the limbs and buttocks ([24 – 27] and reviewed in
of the importance of PPARγ’s role in maintaining the integ-
[15]), severe insulin resistance, diabetes and hypertension
rity and function of the mature adipocyte has been recently
[28]. Although these studies provide direct genetic evidence
shown through the selective ablation of total PPARγ in
of a link between PPARγ action and the regulation of mam-
adipocytes of adult mice [35]. PPARγ-deficient mature
malian glucose homeostasis, it remains uncertain whether
adipocytes die within a few days, but are replaced days later
the profound effects on insulin resistance observed in these
with newly differentiated PPARγ-expressing adipocytes.
individuals is only a manifestation of reduced adipose tissue
Thus PPARγ is essential for the in vivo survival of mature
mass or whether other direct effects of PPARγ action on
adipocytes and hence PPARγ antagonists are potentially
insulin signalling are impaired. In contrast to the individuals
usefully to reduce obesity acutely [35]. The PPARγ gene
with loss of function mutations, a rare Pro115Gln substitu-
encodes two isoforms that are generated by the use of alternate
tion renders PPARγ constitutively active and carriers of this
promoters and differential splicing sites [36,37]. PPARγ2
mutation are obese but remain insulin sensitive [29]. PPARγ
has an additional 30 amino acids in its NH terminal, which
activity in humans corresponds directly to adipose mass, and
is thought to be why PPARγ2 is more effective in gene acti-
not necessarily with insulin sensitivity (Fig. 2), suggesting
vation compared to PPARγ1 [38]. Until recently, the relative
that only part of PPARγ’s effects on glucose homeostasis is
contribution of the two PPARγ isoforms for adipogenesis
dependent on white adipose tissue (WAT). in vivo remained unknown, but in vitro studies suggest thatPPARγ2 is better suited to adipogenesis [39,40]. Studies inmice selectively disrupted in adipose PPARγ2 expression
Mouse genetic variants
demonstrated reduced levels of adipose tissue, specificallyWAT and were insulin insensitive [41]. The lack of a com-
Genetic manipulations of PPARγ in the mouse began with
plete absence of fat indicates that PPARγ1 alone is able to
the generation of a PPARγ–/– mouse using conventional gene
drive the development of adipose tissue but that PPARγ2
targeting strategies. Unfortunately, these mice die in utero
plays the dominant role in adipogenesis. The presence of
resulting from major placental and cardiac defects and
impaired insulin sensitivity in the face of normal glucose
although a PPARγ–/– animal could be rescued by tetraploid
tolerance in these mice was hypothesized to be a conse-
aggregation, this mouse died within days as a result of severe
quence of reduced levels of plasma leptin and adiponectin
lipodystrophy [30]. The lipodystrophy in this PPARγ–/– mouse
2005 Blackwell Publishing Ltd, European Journal of Clinical Investigation, 35, 82– 92
PPARγ, the more the merrier? 85 Table 1 A comparison of the main PPARγ tissue specific knockout mice models
The different mouse models have various age and diet-dependent responses, thus for simplicity the above descriptions are for the adult
phenotype in the postprandial state on a chow diet, at the age indicated.
Abbreviations: N/D (not determined), TG (triglcyerides), FFA (free fatty acids), apoE (apolipoprotein E), LDL (low density lipoprotein).
insulin-sensitizing effects of TZDs on muscle are indirect
PPARγ in the liver and muscle
and that muscle PPARγ is not involved in insulin sensing
Although PPARγ is predominantly expressed in the adipo-
[44]. These findings are consistent with the observations in
cyte, both the skeletal muscle and the liver express small
the PPARγhyp/hyp mouse model that shows adipose tissue
but significant levels of PPARγ and furthermore, are tissues
PPARγ expression is crucial for the insulin-sensitizing effects
that are important in glucose and fuel homeostasis. There-
of TZDs, as TZD treatment of these mice only ameliorated
fore, tissue-specific knockout models were created to define
glucose intolerance but not insulin resistance [33]. In con-
PPARγ’s function in the muscle and liver. Consistent with
trast to Norris et al., Hevener et al. reported that mice lack-
the adipose tissue-specific PPARγ knockout models, lipid
ing PPARγ specifically in the muscle develop severe muscle
balance was also significantly altered when either muscle or
insulin resistance and as a result are hyperinsulinemic,
liver PPARγ was obliterated. Deletion of PPARγ in the livers of
glucose intolerant and hypertriglyceridemic. Because TZD
two mouse models with significant steatosis (leptin-deficient
treatment of these mice did not augment the insulin-
ob/ob or lipodystrophic A-ZIP/ F-1 mice) reduced the liver
stimulated glucose disposal by the muscle the authors con-
triglyceride content, although it elevated serum FFAs
cluded that TZD treatment did not enhance muscle insulin
and lipoproteins and induced insulin resistance, illustrating
sensitivity and therefore muscle PPARγ is a direct target of
PPARγ’s role in liver lipogenesis [42,43]. Because in the
TZDs [45]. Most studies however, suggest that PPARγ in
absence of WAT, the ability of TZD (thiazolidinedione)
the muscle is more responsible for coordinating the use of
treatment to lower triglycerides and glucose was dependent
energy rather than directly controlling glucose homeostasis
on liver PPARγ, the general consensus is that in the absence
or responses to insulin [33,34,42 – 44], validating the con-
of WAT, liver PPARγ participates in both fat regulation and
cept that WAT is predominantly responsible for the insulin-
glucose homeostasis [42] but in the presence of WAT, the
sensitizing effects of PPARγ. PPARγ in the WAT therefore,
impact of PPARγ in the liver on glucose homeostasis is
may not only be the master regulator of adipogenesis in vivo
minimal. The role of PPARγ in the muscle is much less obvi-
but also a driving force of glucose and lipid homeostasis.
ous in light of two independent reports of muscle-specific
The above mouse models have helped us to realize that
PPARγ knockout mouse models, which are essentially oppo-
when PPARγ is absent in any of the three tissues, whole-body
site [44,45]. Norris et al. report that muscle-specific PPARγ
lipid homeostasis and insulin sensitivity are significantly
knockout mice have normal glucose homeostasis and insulin
altered. This is a fascinating observation considering that
levels but have reduced hepatic insulin sensitivity, suggested
PPARγ expression levels vary greatly among these tissues
to be a consequence of the increased WAT mass. The inef-
with adipose tissue having the highest PPARγ expression
ficient use of lipid as fuel by the muscle explained the shunt
levels and the liver and muscle very little. The resulting
of lipid to the adipocyte and the enhanced adiposity. As dis-
repartitioning of lipids that occurs in these mouse models
ruption of muscle PPARγ did not block the beneficial effects
has also unveiled the presence of a complex network of cross
of TZDs on glucose homeostasis, it was concluded that the
talk between the liver, adipose and muscle that is essential
2005 Blackwell Publishing Ltd, European Journal of Clinical Investigation, 35, 82– 92
in order to maintain energy balance. This balance is in part
type 2 diabetic patients (reviewed in [55] and [56]). TZDs
achieved by the adaptation of PPARγ in the nontargeted
increase WAT mass, redistribute it from visceral to sub-
tissues and by the other PPAR isoforms, PPARα and β/δ,
cutaneous deposits and induce the appearance of small,
which enhance fatty acid oxidation to minimize hyperlipi-
newly differentiated adipocytes (reviewed in [57]). TZDs,
daemia and the consequential insulin resistance.
furthermore have an impact on the production of FFAs andthe secretion of several adipokines such as TNFα, leptin,resistin, and adiponectin and can also via this way, affect
PPARγ in other mouse tissues
insulin signalling in other tissues (reviewed in [58]). Exciting
In contrast to adipose tissue, muscle or liver, the deletion of
is the development of a new class of high-affinity tyrosine-based
PPARγ in the pancreas did not result in a metabolic phenotype,
receptor agonists, exemplified by farglitazar, that acts by
but it underscored the antiproliferative role of PPARγ [46].
releasing the corepressor, silencing mediator of retinoic acid
In comparison to adipocyte PPARγ, macrophage PPARγ
and thyroid hormone receptors (SMRT) from PPARγ [59].
has a similar function in that it regulates lipid homeostatic
These SMRT releasers are extremely potent and efficacious
genes including LPL and CD36. In macrophage-specific
PPARγ agonists that can even promote wild-type levels
PPARγ deficient mice lipid homeostasis in the arterial wall
of transcriptional activation by the PPARγ mutants Val290Met
was significantly impaired and the development of athero-
and Pro467Leu mutants, both of which respond poorly to
classical TZD treatment [59]. These data underscore theimportance of corepressor release in attaining nuclear receptortranscriptional responses [59,60]. Human PPARγ mutations in the mouse
Whereas potent and efficacious PPARγ activation is invari-
The knowledge obtained from the tissue specific deletions of
ably associated with increased fat mass, PPARγ antagonism
PPARγ is further refined by the generation of mouse models
is neutral or even reverses weight gain. This principle was
carrying specific PPARγ mutations such as the knock-in
illustrated by the binding of the partial agonist FMOC-L-
of alanine at position 112 (S112A). This mutation, which
Leu to PPARγ, which induces the differential recruitment
renders PPARγ constitutively active (like the human Pro115Gln
of coregulators to PPARγ such that glucose levels are still
mutation) preserves insulin sensitivity during diet-induced
lowered but there is no weight gain [54]. Alternatively, the
obesity [49] as a result of smaller fat cells, elevated serum
partial PPARγ agonists, such as NC-2100 [61] or MCC-
adiponectin and reduced FFA levels. Thus the phosphor-
555 [62], also show little effect on adipocyte differentiation,
ylation state of PPARγ modulates insulin sensitivity suggesting
but have remarkable antidiabetic activities. Partial inhibition
that compounds designed to modulate PPARγ phosphor-
of either PPARγ or its heterodimerization partner the retin-
ylation may selectively enhance insulin sensitivity without
oid X receptor (RXR) by antagonists [63 – 65] also improved
increasing weight gain. Another mouse model that expresses
insulin sensitivity, consistent with human and mouse genetic
the analogue of a human dominant negative PPARγ muta-
studies. Whether there are PPARγ-independent effects of
tion, Pro467Leu [50] tackled the dogma that hypertension
TZDs which can account for some of these differences is
is a consequence of the insulin resistance in the wake of
currently under intense investigation [7,66,67].
PPARγ deficiency and lipodystrophy [51]. The PPARγ
Collectively, the mouse models of ablated PPARγ expres-
Pro467Leu mice develop severe hypertension despite mild
sion in metabolic tissues, the human mutational analysis
fat redistribution and minimal insulin resistance. This
and these pharmacological studies demonstrate that PPARγ
uncoupling between lipodystrophy, insulin resistance and
activity corresponds directly to adiposity in a linear fashion
hypertension, implies direct modulation of blood pressure
(Fig. 2A). However, unlike PPARγ’s relationship with fat mass,
by PPARγ possibly through regulating the renin angiotensin
PPARγ activity is not linearly related to insulin sensitivity,
system activity in adipose tissue [50,51]. This hypothesis
as inactivation and activation of PPARγ can both enhance
supports the decrease in blood pressure observed with TZD
insulin sensitivity (Fig. 2B). This nonlinear relationship indi-
treatment and emphasizes the continued value of using
cates that insulin sensitivity is an integrated effect achieved
natural mutations to understand receptor function and
predominantly by modulating PPARγ actions within the
adipose tissue with effects on adipokine secretion and lipidstorage in addition to other tissue-specific PPARγ responses,as revealed by tissue-specific PPARγ obliterated models. Pharmacological studies
PPARγ binds multiple ligands that can modulate its activityand induce a full spectrum of receptor activities from full
PPARγ and bone homeostasis
inhibition (antagonist) to activation (agonist) (Fig. 2). PPARligands have been generated with varying degrees of effects
In terms of a PPARγ-driven thrifty gene response, physical
which can be attributed to their ability to either differentially
activity and food procurement are inextricably linked in that
recruit cofactors [54] or to selectively activate or inactivate
physical activity is required to obtain food before the energy
PPARγ tissue-specific manner (SPPARMS). Full PPARγ
in the food can be used or stored. Thus, it is plausible that
agonists, such as the first generation TZDs, improve insulin
concomitant with PPARγ’s role in securing a constant sup-
sensitivity, glucose tolerance, and the lipidemic profile in
ply of substrate to fuel muscle contraction and brain activity,
2005 Blackwell Publishing Ltd, European Journal of Clinical Investigation, 35, 82– 92
PPARγ, the more the merrier? 87
it also regulates bone mass to provide the physical strength
and gelatinase B (MMP-9) by a DNA binding-independent
required in procuring the next meal. PPARγ’s influence on
mechanism involving negative interference with the trans-
osteogenesis and bone homeostasis was first suggested
cription factors AP-1, NF-κB and STAT-1. Furthermore,
by the single silent nucleotide mutation polymorphism of
PPARγ ligands inhibited the phorbol ester-induced produc-
PPARγ that was associated with lower bone mineral density
tion of proinflammatory cytokines, TNFα, IL-1 and Il-1α
[68] and higher leptin levels [69]. Studies using natural and
[78]. These inhibitory effects would be expected to be bene-
synthetic PPARγ agonists [70,71] also demonstrated adverse
ficial in the context of atherosclerosis, however, it was orig-
effects on bone formation in mice. Whether these effects
inally uncertain whether the 15d-PGJ - and TZD-mediated
reflected PPARγ-dependent or PPARγ-independent effects
inhibition of the synthesis of these pro-inflammatory genes
of these PPARγ agonists remained, however, elusive until
was a PPARγ-dependent effect [74]. Studies in PPARγ-null
heterozygous PPARγ-deficient mice were reported to have
macrophages demonstrated that contrary to the initial
enhanced bone mass as a result of increased osteoblasto-
beliefs, PPARγ is neither essential for macrophage differ-
genesis [72]. But which PPARγ-expressing tissue contributed
entiation or for mature macrophage functions such as
to enhanced bone formation? This question was answered
pro-inflammatory cytokine production [79,80].
in the severely lipodystrophic PPARγhyp/hyp mouse model,
In addition to the inflammatory responses mediated
which do not express PPARγ1 and PPARγ2 in WAT [73].
by PPARγ ligands they may also influence atherosclerosis
The specific absence of PPARγ in fat robustly increased
through regulating macrophage lipid and lipoprotein
bone mass as it favoured mesenchymal stromal precursor
metabolism. Macrophage uptake of atherogenic lipoproteins
cells to undergo osteogenic differentiation rather than adipo-
within the arterial wall results in cholesteryl ester deposition
genic differentiation. The absence of PPARγ in adipocytes
and foam cell formation, which are hallmarks of early and
also limited their capacity to secrete antiosteogenic-signalling
late atherosclerosis. PPARγ targets both lipoprotein uptake
factors, including leptin, further enhancing the bone
and cholesterol efflux, two competing processes involved in
phenotype [73]. In addition, the strongly enhanced bone
macrophage lipid homeostasis. PPARγ activation induces
mass consequentially reduced the bone marrow cavity and
expression of the scavenger receptor CD36, thereby pro-
hematopoiesis. Bone marrow hematopoiesis was compen-
moting oxidised low density lipoprotein (oxLDL) uptake
sated for by extramedullary hematopoiesis in the spleen
and formation of foam cell. In addition to the acquisition
[73]. If these data obtained in the mouse models can be
of cholesterol, however, macrophage uptake of oxLDL
extrapolated to humans, inhibition of PPARγ activity could
provides the cell with naturally occurring PPARγ ligands,
be an interesting strategy to combat osteoporosis. It also
thereby promoting further PPARγ activation and CD36 up-
warrants careful following of T2DM patients treated
regulation. Such a feed-forward cycle predicts that PPARγ
with PPARγ agonists to detect eventual development of
is predominantly pro-atherogenic [75,81]. However, subse-
quent studies in mice treated with PPARγ or RXR ligandsdemonstrated reduced atherosclerosis [82 – 85]. This anom-aly was resolved when PPARγ activation was shown to pro-mote the removal of cholesterol from macrophages through
PPARγ and atherosclerosis
enhancing the cholesterol efflux mediated by the ATP-bindingcassette transporter A1 (ABCA1). This stimulates HDL
In addition to the above thrifty activities of PPARγ in WAT,
formation (high-density lipoprotein) and reverse cholesterol
PPARγ activity in alternate cells such as macrophages might
transport. The expression of the ABCA1 is tightly regulated by
also be beneficial from an evolutionary perspective as it
cellular cholesterol content through the oxysterol-dependent
could favour the innate immune response [74]. As PPARγ
activation of another nuclear receptor, the liver X receptor
is also expressed in endothelial and smooth muscle cells
(LXR). PPARγ has been shown to activate ABCA1 expres-
there has been a push to understand the role of PPARγ in
sion indirectly via enhanced transcription of LXR [47,83]
the vasculature, in order to unravel the complex pathophysi-
and possibly through coupled up-regulation of LXR ligand
ologic alterations that relate insulin resistance and metabolic
production; as PPARγ has been recently demonstrated to
perturbations to tissue injury in the blood vessel leading to
up-regulate CYP27 expression and consequently the pro-
atherosclerosis. An important outstanding question is
duction of the oxysterol, 27-hydroxycholesterol [86]. PPARγ
whether PPARγ influences the risk of myocardial infarction
and LXR cooperate to modulate other lipid regulating
genes. Conditional disruption of PPARγ in mice in addition
One way PPARγ may influence cardiovascular disease
to lowering the expression of ABCA1 and ABCG1 also
development is through modulating arterial macrophage
lowered the expression of apoE, CD36, LXRα and LPL
inflammation, lipid and lipoprotein metabolism. PPARγ
genes [48]. Given that both of these nuclear receptors are
is expressed in monocytes and up-regulated during their
activated by lipid components of oxLDL, PPARγ and LXR
differentiation into macrophages [75]. In fact, the extent of
actually comprise a cascade that coordinates a response to
macrophage differentiation or activation has been linked to
oxLDL uptake [87]. Whether this response is pro or antia-
the extent of PPARγ expression [76]. PPARγ ligands oppose
therogenic will depend on their net effect on cellular processes
several events that occur during macrophage activation.
mediating lipid uptake, cholesterol efflux and inflammation.
Ricote et al. [77] demonstrated that natural and synthetic
These data emphasize the potential involvement of
PPARγ ligands inhibited IFNγ-induced expression of iNOS
PPARγ in the pathogenesis of atherosclerosis, which was
2005 Blackwell Publishing Ltd, European Journal of Clinical Investigation, 35, 82– 92
underscored in humans by the association of the PPARγPro12Ala polymorphism with a protection from coronaryheart disease [88] and more recently with reduced carotidintimal medial thickness [89]. The efficacy of PPARγ ago-nists to reduce atherosclerosis in mice [82,83] and to exertvasculo-protective effects in humans [90 – 93] are attributedto both its effects on inflammation and cholesterol effluxthat can be receptor-dependent and receptor-independent[66,67,79,80]. Further studies are required to define howmuch of these effects are the result of PPARγ’s beneficial sys-temic metabolic effects vs. its vascular and immune effects,which themselves might be indirect and mediated via LXR. It is also interesting to speculate that the increased frequencyof atherosclerosis could be a PPARγ-driven maladaptedmacrophage response, which occurs when the inherentbeneficial effects (stimulation of the innate immune responseand cholesterol efflux) are overwhelmed by the pro-
Figure 3 Energy metabolism and longevity compared under
atherogenic effects (increased oxLDL uptake). Such condi-
conditions of caloric excess and caloric restriction. Compared to
tions are probably created by the chronic overload of lipids
caloric excess, caloric restriction decreases energy levels leading to
that is associated with our current affluent lifestyle.
activation of a signalling cascade to enhance longevity. Decreased glucose intake by the cell reduces the flow of carbon through the glycolytic pathway and thus decreases glycolytic-derived NADH and the conversion of ADP to ATP. Signalling by the insulin /
PPARγ and longevity
insulin-like growth factor 1 (IGF-1) is attenuated under these conditions, which allows for forkhead transcription factor (FOXO)-
From an evolutionary perspective, the thrifty response
induced stress resistance, cell cycle arrest and apoptosis (antiaging). Sirtuin 1 (Sirt1), a NAD+-regulated chromatin deacetylase,
clearly favours survival when food supply is limited. Caloric
prolongs lifespan in response to caloric restriction in lower
restriction, meaning a diet that is low in calories without
organisms. Sirt1 mediates these effects by deacetylating FOXO3
undernutrition has, however, also been shown to extend
and /or FOXO4, thus attenuating FOXO-induced apoptosis but
mammalian lifespan [94 – 96] (Fig. 3). The beneficial effects
potentiating FOXO-induced cell-cycle arrest [106 –108]. Extremes
of caloric restriction are associated with altered metabolism,
in fat mass are inversely related to lifespan, and PPARγ has recently
particularly reduced metabolic rate and oxidative stress,
been implicated in influencing longevity. Sirt1 may modulate
decreased fat mass, body temperature and fasting glucose
PPARγ target genes and ultimately influence energy expenditure
in addition to improved insulin sensitivity and altered
and fat storage. Dotted lines represent hypothesized effects and the
neuroendocrine and sympathetic nervous system (reviewed in
size of arrow or words corresponds to the importance of the effect.
[97]). One major genetic pathway identified to regulate thelifespan of Caenorhabditis elegans, which is highly conservedamong vertebrates and invertebrates is the insulin and / or
FOXO [104]. Caloric restriction increases Sirt1 expression
insulin-like growth factor-1 (IGF-1) signalling pathway
in several tissues in the rat including the brain, liver, kidney
(IIS) (reviewed in [98]). Reduced signalling through the IIS
and visceral fat pads, a response attenuated in the presence
pathway by mutations in Daf-2, the insulin receptor homo-
of insulin or IGF-1 [105]. Sirt1 may modulate longevity in
logue in C. elegans, can extend lifespan and this response relies
mammals by tipping the balance from cell death towards
on the presence of the C. elegans homologue of the forkhead
cell survival. Sirt1 achieves this effect by regulating the
transcription factors, Daf-16 (FOXO1-3 in mammals)
activity of at least three classes of mammalian damage
([99] and reviewed in [100]). FOXO transcription factors
responsive factors. First, Sirt1 deacetylates the p53 protein
play a key role in transmitting insulin signalling downstream
at lysine 382, thereby inactivating p53-mediated transcrip-
of protein kinase B, which inhibits FOXO activity through
tion and apoptosis. Second, Sirt1 deacetylates the DNA
phosphorylation and nuclear exclusion (Fig. 3). However,
repair factor Ku70, causing it to sequester the proapoptotic
under activating conditions (as seen in cases of decreased
factor Bax away from mitochondria, thereby inhibiting stress-
IIS) FOXO proteins move to the nucleus and regulate genes
induced apoptotic cell death; and finally Sirt1 deacetylates
involved in glucose metabolism, cell cycle regulation,
FOXO3 and /or FOXO4, thus attenuating FOXO-induced
apoptosis and oxidative stress responses. The ultimate
apoptosis but potentiating FOXO-induced cell-cycle arrest
consequence being increased stress resistance, a major hall-
mark of caloric restriction [101 – 103].
Adipose tissue is consistently implicated as a critical tissue
A second longevity regulatory gene is the evolutionarily
in mediating extension of lifespan by altering the IIS path-
conserved NAD+-regulated histone deacetylase silent
way as demonstrated by the extended lifespan in the fat
information regulator (Sir2) (human orthologue Sirt1).
specific insulin receptor knockout mice [99]. This is in line
Sirt1 promotes survival in yeast, C. elegans and mammals
with two recent observations showing that limited expression
in response to food scarcity and this requires the presence of
of the FOXO proteins in the Drosophilia fat body and brain
2005 Blackwell Publishing Ltd, European Journal of Clinical Investigation, 35, 82– 92
PPARγ, the more the merrier? 89
or its overexpression in the Drosophilia fat body is sufficient to
uncontrolled cell proliferation and cancers. However, these
mediate its effects on life span [109,110]. However, repres-
adaptations in the context of our current affluent lifestyles
sion of adipogenesis and fat retention in relation to caloric
that afford excessive exposure to natural PPARγ ligands,
restriction was also recently explained by the inhibition
throw this once tightly regulated system into a metabolic
of PPARγ. Sirt1 was demonstrated to repress PPARγ and
turmoil causing the so-called metabolic syndrome. This
hence activation of fat storage genes, by docking to the
metabolic syndrome encompasses all of today’s most prev-
PPARγ corepressor NCoR and SMRT [111]. In Sirt1+/–
alent diseases including obesity, T2D, and atherosclerosis.
mice, mobilization of fatty acids from white adipocytes upon
Collectively our current knowledge suggests that modulating
fasting was compromised, supporting Sirt1 and PPARγ
(or inhibiting) PPARγ activity, rather than activating it, will be
inactivation as the molecular pathway connecting caloric
the preferred therapeutic strategy to treat metabolic disorders,
restriction to life extension in mammals [111]. Consistent
as this will improve glucose homeostasis, yet prevent
with this mouse model is the fact that the hypomorphic
human Pro12Ala genetic variation in PPARγ was reportedto be associated with increased longevity [21]. Thus PPARγmay represent another longevity regulatory gene.
Two major outstanding questions include how caloric
Acknowledgements
restriction stimulates Sirt1 activity and whether a caloricrestriction mimetic can be developed as a prolongevity strat-
This work was supported by grants from CNRS, INSERM,
egy. With respect to the first dilemma, two hypotheses have
Hopitaux Universitaires de Strasbourg, EU, EMBO and
been proposed to explain caloric restriction and Sirt1 acti-
NIH. We thank the members of the Auwerx Laboratory for
vation: 1) by depleting nicotinamide, an inhibitory product
of Sirt1 itself or 2) by increasing the NAD+/ NADH ratio. Presently, this topic is still under intense debate, however,one recent study disfavours the later hypothesis by demon-strating that under aerobic conditions, the steady-state levels
References
of NAD+ do not fluctuate greatly and its fluctuations duringcaloric restriction negatively correlate with Sirt1 activity
1 Mann NJ. Paleolithic nutrition: what can we learn from the
[112]. With respect to the second question, whether or not
past? Asia Pac J Clin Nutr 2004;13:S17 –23.
pharmacological interventions can be designed to produce
2 Auwerx J. PPARγ, the ultimate thrifty gene. Diabetologia
the same prolongevity effects that caloric restriction pro-
1999;42:1033 – 49.
vides but without substantially reducing caloric intake
3 Rosen ED, Spiegelman BM. PPARγ: a nuclear regulator of
awaits the confirmation that the response to caloric restric-
metabolism, differentiation, and cell growth. J Biol Chem
tion in humans parallels that observed in rodents [113].
2001;276:37731 – 4.
4 Burdge GC, Jones AE, Frye SM, Goodson L, Wootton SA.
However, logical candidates for caloric restriction mimetics
Effect of meal sequence on postprandial lipid, glucose and
would be compounds manipulating the IIS pathway or Sirt1
insulin responses in young men. Eur J Clin Nutr
activity, such as the Sirt1 agonist resveratrol. Resveratrol has
2003;57:1536 – 44.
been shown to extend life span in yeast, nematodes, and files
5 Friedman JM. Modern science versus the stigma of obesity.
in a Sirt1- and caloric restriction-dependent manner [114]. Nat Med 2004;10:563 – 9.
Whether PPARγ modulators or antagonists are potentials
6 Sanders TA. Dietary fat and postprandial lipids. Curr
awaits their further characterization in this pathway. Atheroscler Rep 2003;5:445 –51.
7 Larsen TM, Toubro S, Astrup A. PPARγ agonists in the
treatment of type II diabetes: is increased fatness commensurate with long-term efficacy? Int J Obes Relat Metab Disord 2003;27:147 – 61. The more the merrier?
8 Fonseca V. Effect of thiazolidinediones on body weight in
patients with diabetes mellitus. Am J Med 2003;115 (Suppl.
In general, we are guided by the principle that more is better.
Translated to the PPARγ field, this leads to the initial devel-
9 Tontonoz P, Hu E, Spiegelman BM. Stimulation of
opment of full PPARγ agonists rather than partial PPARγ
adipogenesis in fibroblasts by PPARγ 2, a lipid-activated
agonists. However, in the case of PPARγ activity, the data
transcription factor. Cell 1994;79:1147– 56.
suggest that moderate levels of PPARγ activation coordinate
10 Ren D, Collingwood TN, Rebar EJ, Wolffe AP, Camp HS.
the evolutionary benefit and adaptive response. In a perfect
PPARγ knockdown by engineered transcription factors:
system, input of energy would equal the output. However,
exogenous PPARγ2 but not PPARγ1 reactivates adipogenesis.
evolutionarily speaking, humans have always been subjected
Genes Dev 2002;16:27 –32.
11 Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K,
to dietary fluctuations as during the times of the hunters
Milstone DS et al. PPARγ is required for the differentiation
and gatherers or during these times of affluent lifestyles.
of adipose tissue in vivo and in vitro. Mol Cell 1999;4:611 – 7.
Initially, the adaptation for efficient energy conservation and
12 Schoonjans K, Staels B, Auwerx J. Role of the peroxisome
storage that enabled survival through periods of food short-
proliferator activated receptor (PPAR) in mediating effects of
ages was beneficial including improved longevity and the
fibrates and fatty acids on gene expression. J Lipid Res
enhanced innate immune response combated infections and
1996;37:907 – 25.
2005 Blackwell Publishing Ltd, European Journal of Clinical Investigation, 35, 82– 92
13 Norman RA, Thompson DB, Foroud T, Garvey WT,
28 Barroso I, Gurnell M, Crowley VE, Agostini M,
Bennett PH, Bogardus C et al. Genomewide search for genes
Schwabe JW, Soos MA et al. Dominant negative
influencing percent body fat in Pima Indians: suggestive
mutations in human PPARγ associated with severe insulin
linkage at chromosome 11Q21 Q22. Am J Hum Genet
resistance, diabetes mellitus and hypertension. Nature
1997;60:166 – 73.
1999;402:880 – 3.
14 Florez JC, Hirschhorn J, Altshuler D. The inherited basis
29 Ristow M, Muller-Wieland D, Pfeiffer A, Krone W, Kahn CR.
of diabetes mellitus. Implications for the genetic analysis
Obesity associated with a mutation in a genetic regulator of
of complex traits. Annu Rev Genomics Hum Genet
adipocyte differentiation. New Engl J Med 1998;339:953 – 9.
2004;4:257 – 91.
30 Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P,
15 Knouff C, Auwerx J. PPARγ calls for activation in moderation.
Chien KR et al. PPARγ is required for placental, cardiac, and
Lessons from genetics and pharmacology. Endocr Rev
adipose tissue development. Mol Cell 1999;4:585 – 95.
2004;25:899 – 918.
31 Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T,
16 Deeb S, Fajas L, Nemoto M, Laakso M, Fujimoto W,
Komeda K et al. PPARγ mediates high-fat diet-induced
Auwerx J. A Pro 12 Ala substitution in the human peroxisome
adipocyte hypertrophy and insulin resistance. Mol Cell
proliferator-activated receptor γ2 is associated with decreased
1999;4:597– 609.
receptor activity, improved insulin sensitivity, and lowered
32 Miles PD, Barak Y, He W, Evans RM, Olefsky JM. Improved
body mass index. Nat Genet 1998;20:284 – 7.
insulin-sensitivity in mice heterozygous for PPARγ deficiency.
17 Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM,
J Clin Invest 2000;105:287– 92.
Vohl MC, Nemesh J et al. The common PPARγ Pro12Ala
33 Koutnikova H, Cock TA, Watanabe M, Houten SM,
polymorphism is associated with decreased risk of type 2
Champy MF, Dierich A et al. Compensation by the
diabetes. Nat Genet 2000;26:76 – 80.
muscle limits the metabolic consequences of lipodystrophy in
18 Masugi J, Tamori Y, Mori H, Koike T, Kasuga M. Inhibitory
PPARγ hypomorphic mice. Proc Natl Acad Sci USA
effect of a proline-to-alanine substitution at codon 12 of
2003;100:14457– 62.
peroxisome proliferator-activated receptor-γ2 on
34 He W, Barak Y, Hevener A, Olson P, Liao D, Le J et al.
thiazolidinedione-induced adipogenesis. Biochem Biophys Res
Adipose-specific peroxisome proliferator-activated receptor γ
Commun 2000;268:178 – 82.
knockout causes insulin resistance in fat and liver but not in
19 Muller YL, Bogardus C, Beamer BA, Shuldiner AR, Baier LJ.
muscle. Proc Natl Acad Sci USA 2003;100:15712 – 7.
A functional variant in the peroxisome proliferator-activated
35 Imai T, Takakuwa R, Marchand S, Dentz E, Bornert JM,
receptor γ 2 promoter is associated with predictors of obesity
Messaddeq N et al. Peroxisome proliferator-activated receptor
and type 2 diabetes in Pima Indians. Diabetes
γ is required in mature white and brown adipocytes for their
2003;52:1864 –71.
survival in the mouse. Proc Natl Acad Sci USA
20 Pisabarro RE, Sanguinetti C, Stoll M, Prendez D. High
2004;101:4543 – 7.
incidence of type 2 diabetes in peroxisome proliferator-
36 Fajas L, Auboeuf D, Raspe E, Schoonjans K, Lefebvre AM,
activated receptor γ2 pro12Ala carriers exposed to a high
Saladin R et al. Organization, promoter analysis and
chronic intake of trans fatty acids and saturated fatty acids.
expression of the human PPARγ gene. J Biol ChemDiabetes Care 2004;27:2251– 4.
1997;272:18779 – 89.
21 Barbieri M, Bonafe M, Rizzo MR, Ragno E, Olivieri F,
37 Fajas L, Fruchart JC, Auwerx J. PPARγ 3 mRNA. A distinct
Marchegiani F et al. Gender-specific association of genetic
PPARγ mRNA subtype transcribed from an independent
variation in peroxisome proliferator-activated receptor
promoter. FEBS Lett 1998;438:55 – 60.
(PPAR) γ2 with longevity. Exp Gerontol 2004;39:1095–100.
38 Werman A, Hollenberg A, Solanes G, Bjorbaek C,
22 Yliharsila H, Eriksson JG, Forsen T, Laakso M, Uusitupa M,
Vidal-Puig A, Flier JS. Ligand-independent activation
Osmond C et al. Interactions between peroxisome
domain in the N terminus of peroxisome proliferator-activated
proliferator-activated receptor-γ2 gene polymorphisms and
receptor γ (PPARγ). J Biol Chem 1997;272:20230–5.
size at birth on blood pressure and the use of antihypertensive
39 Saladin R, Fajas L, Dana S, Halvorsen YD, Auwerx J,
medication. J Hypertens 2004;22:1283 – 7.
Briggs M. Differential regulation of peroxisome proliferator
23 Pihlajamaki J, Vanhala M, Vanhala P, Laakso M. The
activated receptor γ1 (PPARγ1) and PPARγ 2 mRNA
Pro12Ala polymorphism of the PPAR γ2 gene regulates weight
expression in early stages of adipogenesis. Cell Growth
from birth to adulthood. Obes Res 2004;12:187 – 90. Differentiation 1999;10:43 – 8.
24 Agarwal AK, Garg A. A novel heterozygous mutation in
40 Ren D, Collingwood TN, Rebar EJ, Wolffe AP, Camp HS.
peroxisome proliferator-activated receptor-γ gene in a patient
PPARgamma knockdown by engineered transcription factors:
with familial partial lipodystrophy. J Clin Endocrinol Metab
exogenous PPARγ2 but not PPARγ1 reactivates adipogenesis.
2002;87:408 –11. Genes Dev 2002;16:27 – 32.
25 Combs TP, Wagner JA, Berger J, Doebber T, Wang WJ,
41 Zhang J, Fu M, Cui T, Xiong C, Xu K, Zhong W et al.
Zhang BB et al. Induction of adipocyte complement-related
Selective disruption of PPARγ2 impairs the development of
protein of 30 kilodaltons by PPARγ agonists: a potential
adipose tissue and insulin sensitivity. Proc Natl Acad Sci
mechanism of insulin sensitization. EndocrinologyUSA 2004;101:10703 – 8.
2002;143:998 –1007.
42 Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L,
26 Hegele RA, Cao H, Frankowski C, Mathews ST, Leff T.
Dietz KR et al. Liver peroxisome proliferator-activated
PPARG F388L, a transactivation-deficient mutant, in familial
receptor gamma contributes to hepatic steatosis, triglyceride
partial lipodystrophy. Diabetes 2002;51:3586 – 90.
clearance, and regulation of body fat mass. J Biol Chem
27 Savage DB, Tan GD, Acerini CL, Jebb SA, Agostini M,
2003;278:34268 – 76.
Gurnell M et al. Human metabolic syndrome resulting from
43 Matsusue K, Haluzik M, Lambert G, Yim SH, Gavrilova O,
dominant-negative mutations in the nuclear receptor
Ward JM et al. Liver-specific disruption of PPARγ in
peroxisome proliferator-activated receptor-γ. Diabetes
leptin-deficient mice improves fatty liver but aggravates
2003;52:910 – 7.
diabetic phenotypes. J Clin Invest 2003;111:737 – 47.
2005 Blackwell Publishing Ltd, European Journal of Clinical Investigation, 35, 82– 92
PPARγ, the more the merrier? 91
44 Norris AW, Chen L, Fisher SJ, Szanto I, Ristow M, Jozsi AC
unique peroxisome proliferator-activated receptor γ-activating
et al. Muscle-specific PPARγ-deficient mice develop increased
properties. J Biol Chem 1998;273:32679 – 84.
adiposity and insulin resistance but respond to
63 Mukherjee R, Hoener PA, Jow L, Bilakovics J, Klausing K,
thiazolidinediones. J Clin Invest 2003;112:608 – 18.
Mais DE et al. A selective peroxisome proliferator-activated
45 Hevener AL, He W, Barak Y, Le J, Bandyopadhyay G,
receptor-gamma (PPARγ) modulator blocks adipocyte
Olson P et al. Muscle-specific PPARγ deletion causes insulin
differentiation but stimulates glucose uptake in 3T3-L1
resistance. Nat Med 2003;9:1491 – 7.
adipocytes. Mol Endocrinol 2000;14:1425 – 33.
46 Rosen ED, Kulkarni RN, Sarraf P, Ozcan U, Okada T,
64 Rieusset J, Touri F, Michalik L, Escher P, Desvergne B,
Hsu CH et al. Targeted elimination of peroxisome
Niesor E et al. A new selective peroxisome proliferator-
proliferator-activated receptor γ in beta cells leads to
activated receptor γ antagonist with antiobesity and
abnormalities in islet mass without compromising glucose
antidiabetic activity. Mol Endocrinol 2002;16:2628 – 44.
homeostasis. Mol Cell Biol 2003;23:7222 – 9.
65 Yamauchi T, Waki H, Kamon J, Murakami K, Motojima K,
47 Chawla A, Boisvert WA, Lee C, Laffitte BA, Barak Y,
Komeda K et al. Inhibition of RXR and PPARγ ameliorates
Joseph SB et al. A PPARγ-LXR-ABCA1 pathway in
diet-induced obesity and type 2 diabetes. J Clin Invest
macrophages is involved in cholesterol efflux and
2001;108:1001 –13.
atherogenesis. Mol Cell 2001;7:161 –71.
66 Rangwala SM, Lazar MA. Peroxisome proliferator-activated
48 Akiyama TE, Sakai S, Lambert G, Nicol CJ, Matsusue K,
receptor γ in diabetes and metabolism. Trends Pharmacol Sci
Pimprale S et al. Conditional disruption of the peroxisome
2004;25:331 – 6.
proliferator-activated receptor γ gene in mice results in
67 Takano H, Hasegawa H, Zou Y, Komuro I. Pleiotropic actions
lowered expression of ABCA1, ABCG1, and apoE in
of PPAR gamma activators thiazolidinediones in
macrophages and reduced cholesterol efflux. Mol Cell Biol
cardiovascular diseases. Curr Pharm Des 2004;10:2779 – 86.
2002;22:2607 –19.
68 Ogawa S, Urano T, Hosoi T, Miyao M, Hoshino S, Fujita M
49 Rangwala SM, Rhoades B, Shapiro JS, Rich AS, Kim JK,
et al. Association of bone mineral density with a polymorphism
Shulman GI et al. Genetic modulation of PPARγ
of the peroxisome proliferator-activated receptor gamma gene:
phosphorylation regulates insulin sensitivity. Dev Cell
PPARγ expression in osteoblasts. Biochem Biophys Res
2003;5:657– 63. Commun 1999;260:122 – 6.
50 Tsai YS, Kim HJ, Takahashi N, Kim HS, Hagaman JR,
69 Meirhaeghe A, Fajas L, Helbecque N, Cottel D, Lebel P,
Kim JK et al. Hypertension and abnormal fat distribution but
Dallongeville J et al. A genetic polymorphism of the
not insulin resistance in mice with P465L PPARγ. J Clin Invest
peroxisome proliferator-activated receptor γ gene influences
2004;114:240 – 9.
plasma leptin levels in obese humans. Hum Mol Genet
51 Hegele RA, Leff T. Unbuckling lipodystrophy from insulin
1998;7:435 – 40.
resistance and hypertension. J Clin Invest 2004;114:163 – 5.
70 Klein RF, Allard J, Avnur Z, Nikolcheva T, Rotstein D,
52 Ryan MJ, Didion SP, Mathur S, Faraci FM, Sigmund CD.
Carlos AS et al. Regulation of bone mass in mice by the
PPARγ agonist rosiglitazone improves vascular function and
lipoxygenase gene Alox15. Science 2004;303:229 – 32.
lowers blood pressure in hypertensive transgenic mice.
71 Rzonca SO, Suva LJ, Gaddy D, Montague DC,
Hypertension 2004;43:661 – 6.
Lecka-Czernik B. Bone is a target for the antidiabetic
53 Dobrian AD, Schriver SD, Khraibi AA, Prewitt RL.
compound rosiglitazone. Endocrinology 2004;145:401 – 6.
Pioglitazone prevents hypertension and reduces oxidative
72 Akune T, Ohba S, Kamekura S, Yamaguchi M, Chung UI,
stress in diet-induced obesity. Hypertension
Kubota N et al. PPARgamma insufficiency enhances
2004;43:48 – 56.
osteogenesis through osteoblast formation from bone marrow
54 Rocchi S, Picard F, Vamecq J, Gelman L, Potier N, Zeyer D
progenitors. J Clin Invest 2004;113:846 – 55. et al. A unique PPARγ ligand with potent insulin-sensitizing
73 Cock TA, Back J, Elefteriou F, Karsenty G, Kastner P,
yet weak adipogenic activity. Mol Cell 2001;8:737 –7.
Chan S et al. Enhanced bone formation in lipodystrophic
55 Schoonjans K, Auwerx J. Thiazolidinediones: an update.
PPARγ (hyp/hyp) mice relocates haematopoiesis to the spleen.
Lancet 2000;355:1008 – 10. EMBO Rep 2004;5:1007 – 12.
56 Yki-Jarvinen H, Thiazolidinediones. N Engl J Med
74 Daynes RA, Jones DC. Emerging roles of PPARs in
2004;351:1106 –18.
inflammation and immunity. Nat Rev Immunol
57 Picard F, Auwerx J. PPARγ and glucose homeostasis. Annu
2002;2:748 –59. Rev Nutr 2002;22:167 – 97.
75 Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM.
58 Moller DE, Kaufman KD. Metabolic syndrome: a clinical and
PPARγ promotes monocyte/macrophage differentiation and
molecular perspective. Annu Rev Med 2005 (in press).
uptake of oxidized LDL. Cell 1998;93:241–52.
59 Agostini M, Gurnell M, Savage DB, Wood EM, Smith AG,
76 Moore KJ, Rosen ED, Fitzgerald ML, Randow F,
Rajanayagam O et al. Tyrosine agonists reverse the molecular
Andersson LP, Altshuler D et al. The role of PPAR-γ in
defects associated with dominant-negative mutations in
macrophage differentiation and cholesterol uptake. Nat Med
human peroxisome proliferator-activated receptor γ.
2001;7:41–7. Endocrinology 2004;145:1527 –38.
77 Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK.
60 Burnstein KL, Luetje CW. Hormone resistance: it’s SMRT
The peroxisome proliferator-activated receptor γ is a
to fight repression. Endocrinology 2004;145:1525 – 6.
negative regulator of macrophage activation. Nature
61 Fukui Y, Masui S, Osada S, Umesono K, Motojima K. A new
1998;391:79 – 82.
thiazolidinedione, NC-2100, which is a weak PPARγ
78 Jiang C, Ting AT, Seed B. PPAR-γ agonists inhibit production
activator, exhibits potent antidiabetic effects and induces
of monocyte inflammatory cytokines. Nature 1998;391:82 – 6.
uncoupling protein 1 in white adipose tissue of KKAy obese
79 Patel L, Charlton SJ, Marshall IC, Moore GBT, Coxon P,
mice. Diabetes 2000;49:759 – 67.
Moores K et al. PPARγ is not a critical mediator of primary
62 Reginato MJ, Bailey ST, Krakow SL, Minami C, Ishii S,
monocyte differentiation or foam cell formation. Biochem
Tanaka H et al. A potent antidiabetic thiazolidinedione with
Biophys Res Commun 2002;290:707 –12.
2005 Blackwell Publishing Ltd, European Journal of Clinical Investigation, 35, 82– 92
80 Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans AJ.
94 Lane MA, de Cabo R, Mattison J, Anson RM, Roth GS,
PPARγ dependent and independent effects on macrophage-
Ingram DK. The Roy Walford legacy: diet restriction from
gene expression in lipid metabolism and inflammation. Nat
molecules to mice to monkeys to man and onto mimetics.
Med 2001;7:48 –52. Exp Gerontol 2004;39:897 – 902.
81 Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM.
95 Sohal RS, Weindruch R. Oxidative stress, caloric restriction,
Oxidized LDL regulates macrophage gene expression through
and aging. Science 1996;273:59 – 63.
ligand activation of PPARγ. Cell 1998;93:229–40.
96 Weindruch RWRL. The retardation of aging and disease by
82 Chen Z, Ishibashi S, Perrey S, Osuga J, Gotoda T, Kitamine T
caloric restriction, Springfiled, IL: CC Thomas; 1988. et al. Troglitazone inhibits atherosclerosis in apolipoprotein
97 Heilbronn LK, Ravussin E. Calorie restriction and aging:
E-knockout mice: pleiotropic effects on CD36 expression and
review of the literature and implications for studies in humans.
HDL. Arterioscler Thromb Vasc Biol 2001;21:372 –7. Am J Clin Nutr 2003;78:361 – 9.
83 Claudel T, Leibowitz MD, Fievet C, Tailleux A, Wagner B,
98 Sell C. Caloric restriction and insulin-like growth factors in
Repa JJ et al. Reduction of atherosclerosis in apolipoprotein
aging and cancer. Horm Metab Res 2003;35:705 – 11.
E knockout mice by activation of the retinoid X receptor. Proc
99 Bluher M, Kahn BB, Kahn CR. Extended longevity in mice
Natl Acad Sci USA 2001;98:2610 –5.
lacking the insulin receptor in adipose tissue. Science
84 Collins AR, Meehan WP, Kintscher U, Jackson S, Wakino S,
2003;299:572 – 4.
Noh G et al. Troglitazone inhibits formation of early
100 Koubova J, Guarente L. How does calorie restriction work?
atherosclerotic lesions in diabetic and nondiabetic low density
Genes Dev 2003;17:313 – 21.
lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc
101 Nemoto S, Finkel T. Redox regulation of forkhead proteins
Biol 2001;21:365 –71.
through a p66shc-dependent signaling pathway. Science
85 Li AC, Brown KK, Silvestre MJ, Willson TM, Palinski W,
2002;295:2450 – 2.
Glass CK. Peroxisome proliferator-activated receptor γ
102 Tran H, Brunet A, Grenier JM, Datta SR, Fornace AJ Jr,
ligands inhibit development of atherosclerosis in LDL
DiStefano PS et al. DNA repair pathway stimulated by the
receptor-deficient mice. J Clin Invest 2000;106:523 – 31.
forkhead transcription factor FOXO3a through the Gadd45
86 Szanto A, Benko S, Szatmari I, Balint BL, Furtos I, Ruhl R
protein. Science 2002;296:530 – 4. et al. Transcriptional regulation of human CYP27 integrates
103 Guarente L, Kenyon C. Genetic pathways that regulate ageing
retinoid, peroxisome proliferator-activated receptor, and liver
in model organisms. Nature 2000;408:255 – 62.
X receptor signaling in macrophages. Mol Cell Biol
104 Blander G, Guarente L. The Sir2 family of protein
2004;24:8154 – 66.
deacetylases. Annu Rev Biochem 2004;73:417 – 35.
87 Repa JJ, Mangelsdorf DJ. The liver X receptor gene team:
105 Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B,
potential new players in atherosclerosis. Nate Med
Kessler B et al. Calorie restriction promotes mammalian cell
2002;8:1243 – 8.
survival by inducing the SIRT1 deacetylase. Science
88 Ridker PM, Cook NR, Cheng S, Erlich HA, Lindpaintner K,
2004;305:390 – 2.
Plutzky J et al. Alanine for proline substitution in the
106 Campisi J. Fragile fugue. p53 in aging, cancer and IGF
peroxisome proliferator-activated receptor gamma-2
signaling. Nat Med 2004;10:231 – 2.
(PPARG2) gene and the risk of incident myocardial infarction.
107 Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL,
Arterioscler Thromb Vasc Biol 2003;23:859 – 63.
Lin Y et al. Stress-dependent regulation of FOXO
89 Temelkova-Kurktschiev T, Hanefeld M, Chinetti G,
transcription factors by the SIRT1 deacetylase. Science
Zawadzki C, Haulon S, Kubaszek A et al. Ala12Ala Genotype
2004;303:2011– 5.
of the peroxisome proliferator-activated receptor γ2 protects
108 Giannakou ME, Partridge L. The interaction between FOXO
against atherosclerosis. J Clin Endocrinol Metab
and SIRT1: tipping the balance towards survival. Trends Cell
2004;89:4238 – 42. Biol 2004;14:408 – 12.
90 de Dios ST, Bruemmer D, Dilley RJ, Ivey ME, Jennings GL,
109 Hwangbo DS, Gersham B, Tu MP, Palmer M, Tatar M.
Law RE et al. Inhibitory activity of clinical thiazolidinedione
Drosophila dFOXO controls lifespan and regulates insulin
peroxisome proliferator activating receptor-γ ligands toward
signalling in brain and fat body. Nature 2004;429:562 – 6.
internal mammary artery, radial artery, and saphenous vein
110 Giannakou ME, Goss M, Junger MA, Hafen E, Leevers SJ,
smooth muscle cell proliferation. Circulation
Partridge L. Long-lived Drosophila with overexpressed
2003;107:2548 – 50.
dFOXO in adult fat body. Science 2004;305:361.
91 Koshiyama H, Shimono D, Kuwamura N, Minamikawa J,
111 Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T,
Nakamura Y. Rapid communication: inhibitory effect of
Machado DO et al. Sirt1 promotes fat mobilization in white
pioglitazone on carotid arterial wall thickness in type 2
adipocytes by repressing PPARγ. Nature 2004;429:771–6.
diabetes. J Clin Endocrinol Metab 2001;86:3452 – 6.
112 Anderson RM, Latorre-Esteves M, Neves AR, Lavu S,
92 Sekiya M, Suzuki J, Watanabe K, Funada J, Otani T,
Medvedik O, Taylor C et al. Yeast life-span extension by
Akutsu H. Beneficial effect of troglitazone, an insulin-
calorie restriction is independent of NAD fluctuation. Science
sensitizing antidiabetic agent, on coronary circulation in
2003;302:2124 – 6.
patients with non-insulin-dependent diabetes mellitus. Jpn
113 Ingram DK, Anson RM, de Cabo R, Mamczarz J, Zhu M,
Circ J 2001;65:487 – 90.
Mattison J et al. Development of calorie restriction mimetics
93 Takagi T, Akasaka T, Yamamuro A, Honda Y, Hozumi T,
as a prolongevity strategy. Ann N Y Acad Sci
Morioka S et al. Troglitazone reduces neointimal tissue
2004;1019:412 – 23.
proliferation after coronary stent implantation in patients with
114 Wood JG, Rogina B, Lavu S, Howitz K, Helfand SL,
non-insulin dependent diabetes mellitus: a serial intravascular
Tatar M et al. Sirtuin activators mimic caloric restriction and
ultrasound study. J Am Coll Cardiol 2000;36:1529 – 35.
delay ageing in metazoans. Nature 2004;430:686 – 9.
2005 Blackwell Publishing Ltd, European Journal of Clinical Investigation, 35, 82– 92
Garantiebedingungen, Sicherheitshinweise & Montageanleitung für Lithium Akkus und Ladegeräte. Es handelt sich um Zubehör der Fa. Tantepaula24 – keine original TP-Teile! Lithium-Eisen-Phosphat (LiFePo4) und Lithium-Nickel-Mangan-Cobalt-Oxid (LiNiMnCoO2) - Akkus Lithium Akkus bedürfen besonderer Sorgfalt und Vorsicht um Risiken zu minimieren und die Lebensdauer der Batterie
Eesti ravimistatistika 2006-2009, lk 30-34 Estonian Statistics on Medicines 2006-2009, pp. 30-34 Diabeediravimite kasutamine Eestis Use of Drugs used in Diabetes Endocrinologist, doctor of medicine, Endocrinology Estonia has not been left untouched by the world-puutumata jäänud ka Eesti. See kajastub wide epidemic of diabetes. It is reflected in the süstitavate ja suukaudset
 PPARγ, the more the merrier? 83
PPARγ, the more the merrier? 83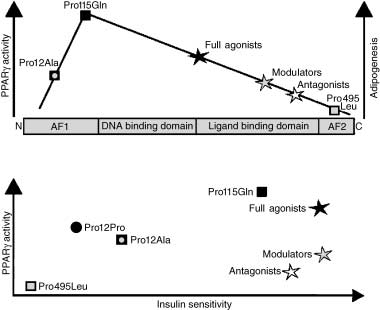 together with the characterization of mice chimaeric forPPARγ–/– ES cells [11], showed the importance of PPARγin adipose tissue development in vivo. Until the past year,determination of the physiological function of PPARγin mice had been limited to studies in the heterozygousPPARγ+/– mice, which was a difficult model to comprehendas these mice are resistant to the obesity and insulin resist-ance that is induced by a high-fat diet [31,32]. To overcomethe embryonic lethality, mice with tissue-specific deletionsof PPARγ have been generated to help elucidate the tissuespecific activities of PPARγ (summarized in Table 1).
together with the characterization of mice chimaeric forPPARγ–/– ES cells [11], showed the importance of PPARγin adipose tissue development in vivo. Until the past year,determination of the physiological function of PPARγin mice had been limited to studies in the heterozygousPPARγ+/– mice, which was a difficult model to comprehendas these mice are resistant to the obesity and insulin resist-ance that is induced by a high-fat diet [31,32]. To overcomethe embryonic lethality, mice with tissue-specific deletionsof PPARγ have been generated to help elucidate the tissuespecific activities of PPARγ (summarized in Table 1).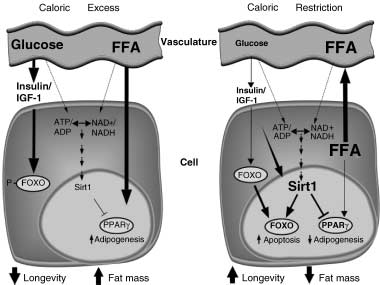 underscored in humans by the association of the PPARγPro12Ala polymorphism with a protection from coronaryheart disease [88] and more recently with reduced carotidintimal medial thickness [89]. The efficacy of PPARγ ago-nists to reduce atherosclerosis in mice [82,83] and to exertvasculo-protective effects in humans [90 – 93] are attributedto both its effects on inflammation and cholesterol effluxthat can be receptor-dependent and receptor-independent[66,67,79,80]. Further studies are required to define howmuch of these effects are the result of PPARγ’s beneficial sys-temic metabolic effects vs. its vascular and immune effects,which themselves might be indirect and mediated via LXR.
underscored in humans by the association of the PPARγPro12Ala polymorphism with a protection from coronaryheart disease [88] and more recently with reduced carotidintimal medial thickness [89]. The efficacy of PPARγ ago-nists to reduce atherosclerosis in mice [82,83] and to exertvasculo-protective effects in humans [90 – 93] are attributedto both its effects on inflammation and cholesterol effluxthat can be receptor-dependent and receptor-independent[66,67,79,80]. Further studies are required to define howmuch of these effects are the result of PPARγ’s beneficial sys-temic metabolic effects vs. its vascular and immune effects,which themselves might be indirect and mediated via LXR.