La tétracycline, connue sous le nom commercial Sumycin, agit en bloquant la fixation de l’ARNt sur la sous-unité 30S ribosomale, interrompant l’élongation de la chaîne protéique bactérienne. Ce mécanisme confère une activité sur un spectre large, incluant bactéries Gram positives, Gram négatives, rickettsies et spirochètes. Sa biodisponibilité digestive varie selon la prise alimentaire et les interactions avec les ions divalents comme calcium et magnésium. Sa diffusion tissulaire est importante, notamment dans les voies respiratoires et génito-urinaires. L’élimination se fait par voie rénale et biliaire. Les effets indésirables incluent photosensibilisation, troubles digestifs et coloration dentaire en cas d’administration précoce. Les guides thérapeutiques mentionnent sumycin prix, en soulignant la nécessité de restreindre son utilisation afin de limiter les résistances acquises.
No job name
Biochemistry 2003, 42, 257-264
LytG of Bacillus subtilis Is a Novel Peptidoglycan Hydrolase: The Major Active
Gavin J. Horsburgh, Abdelmadjid Atrih, Michael P. Williamson, and Simon J. Foster*
Department of Molecular Biology and Biotechnology, UniVersity of Sheffield, Sheffield, S10 2TN, United KingdomReceiVed July 29, 2002; ReVised Manuscript ReceiVed October 21, 2002
ABSTRACT: LytG (YubE) of Bacillus subtilis is a novel 32 kDa autolysin produced during vegetativegrowth under the control of EσA RNA polymerase. Muropeptide analysis of vegetative cells of B. subtilisrevealed LytG to be the major glucosaminidase responsible for peptidoglycan structural determinationduring vegetative growth. Overexpression and purification of LytG allowed its biochemical characterization. Despite sequence homology suggesting muramidase activity, LytG is a novel glucosaminidase withexoenzyme activity and may form part of a novel family of autolysins. It is involved in cell division,lysis, and motility on swarm plates.
Peptidoglycan is essential for the maintenance of cellular
no change in growth, division or sporulation (20). Although
viability and shape determination for most eubacteria. It is
Ortiz et al. (19) suggest the production of a similar exo- -
a dynamic structure continually being synthesized, modified,
N-acetylglucosaminidase at low levels in B. subtilis 168, no
and hydrolyzed to allow for cell growth and division.
further evidence of its existence has been shown.
Bacteria produce a complement of autolysins capable of
Renaturing SDS-PAGE analysis and the genome se-
hydrolyzing the peptidoglycan structure of their own cell wall
quence of B. subtilis have revealed the presence of multiple
(1). These autolysins have been implicated in several
putative novel autolysins, which have begun to be character-
important cellular functions, including differentiation, cell
ized (21-25). The complex compensatory nature of the
lysis, cell wall growth and turnover, cell separation, com-
autolysins means that it is important to identify and analyze
petence, motility, and antibiotic-induced lysis (2-6). Bacillus
the total complement of these enzymes to determine their
subtilis 168 has two major autolysins expressed during the
individual and combined roles. One of the putative novel
vegetative phase of growth, a 50 kDa N-acetylmuramyl-L-
autolysins identified by sequence homology is a 32kDa
alanine amidase (LytC) and a 90 kDa N-acetylglucosamini-
possible muramidase (25), encoded by the lytG (formerly
dase (LytD) (7, 8). Both enzymes are dispensable for growth
yubE) gene, and shows greatest homology to the autolysins
but have often mutually compensatory roles in cell wall
AcmA of Lactococcus lactis and AlyS of Enterococcus hirae.
turnover, motility, and cell separation (9-13). Expression
The lytG gene is situated in a possible bi-cistronic operon
of lytC and lytD is mainly under the control of the sigma
with the downstream gene yubF, which encodes a putative
factor, σD,1 which controls the flagellar, chemotaxis and
protein of unknown function. In this study, we have
motility regulon (12, 14, 15).
characterized LytG as a novel exoglucosaminidase with a
As well as the discovery of the endo- -N-acetylglu-
cosaminidase (LytD) in B. subtilis 168 an exo- -N-acetyl-glucosaminidase has been characterized from B. subtilis B(16-19). This exo- -N-acetylglucosaminidase has a molec-
EXPERIMENTAL PROCEDURES
ular weight of about 75 kDa and an optimum pH of 5.9. Amutant lacking the exo- -N-acetylglucosaminidase showed
Bacterial Strains, Plasmids, and Growth Conditions. All
B. subtilis strains and plasmids used in this study are shown
in Table 1. Unless otherwise stated B. subtilis strains were
This research was funded by the BBSRC and the Royal Society.
* Corresponding author. Mailing address: Department of Molecular
grown in nutrient broth (Oxoid) at 37 °C with shaking (250
Biology and Biotechnology, University of Sheffield, Firth Court,
rpm) or on nutrient agar plates at 37 °C. Plasmids were
Western Bank, Sheffield, S10 2TN, United Kingdom. Phone: 44 114
constructed in, and prepared from, Escherichia coli strain
222 4411. Fax: 44 114 272 8697. E-mail: s.foster@sheffield.ac.uk.
1 Abbreviations, RP-HPLC, reverse phase high-pressure liquid chro-
DH5R grown in Luria-Bertani (LB) broth or on LB agar at
matography; σ, sigma factor; COSY, correlated spectroscopy; TOCSY,
37 °C. When appropriate, chromosomal drug resistance
total correlated spectroscopy; ROESY, rotating frame nuclear Over-
markers in B. subtilis were selected with kanamycin (10 µg
hauser effect spectroscopy; EDTA, ethylenediaminetetraacetic acid; WT,
mL-1), erythromycin (1 µg mL-1), lincomycin (25 µg mL-1),
wild type; CWBP, cell wall binding protein; MurNAc, N-acetylmuramicacid; GlcNAc, N-acetylglucosamine; A2pm, meso-diaminopimelic acid.
spectinomycin (100 µg mL-1), phleomycin (0.3 µg mL-1),
258 Biochemistry, Vol. 42, No. 2, 2003trpC2 metB5 xin-1 SP (s) lytG::pMUTIN4, EmrtrpC2 metB5 xin-1 SP (s) lytC::bletrpC2 metB5 xin-1 SP (s) lytD::spctrpC2 metB5 xin-1 SP (s) sigD::pLM5 CmrtrpC2 metB5 xin-1 SP (s) lytC::ble lytD::spctrpC2 metB5 xin-1 SP (s) lytC::ble lytD::spc sigD::pLM5 CmrtrpC2 metB5 xin-1 SP (s) lytG::pMUTIN4, Emr lytC::bletrpC2 metB5 xin-1 SP (s) lytG::pMUTIN4, Emr lytD::spctrpC2 metB5 xin-1 SP (s) lytG::pMUTIN4, Emr lytC::ble lytD::spctrpC2 metB5 xin-1 SP (s) lytG::pMUTIN4, Emr lytC::ble lytD::spc sigD::pLM5 CmrtrpC2 metB5 xin-1 SP (s) yubF::kantrpC2 metB5 xin-1 SP (s) yubF::kan lytC::bletrpC2 metB5 xin-1 SP (s) yubF::kan lytC::ble lytD::spcsupE44∆ lacU169 (Φ80 lacZ ∆M15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1
pUC119 with 2.25 kb BamHI-SacI fragment containing yubF gene with an
pUC119 with 2.25 kb BamHI-SacI fragment containing yubF gene with a kanamycin
cassette inserted in the engineered XhoI site
pMUTIN4 containing a 300 bp HindIII-BamHI insert carrying part of the yubE gene
pET24d containing lytG gene with its signal sequence removed and C-terminal
a Arrows (>) indicate transformation of recipient strain with donor chromosomal DNA. b Bacillus Genetic Stock Center, Ohio State University,
or chloramphenicol (5 µg mL-1). In E. coli, plasmids were
and also 33 bp into the yubF gene. A kanamycin cassette
selected with ampicillin (50 µg mL-1).
was excised from pDG782 (29) by digestion with XhoI and
Construction of Mutants Insertionally InactiVated in lytGSalI and then ligated with pGJH170. The resultant plasmid,
and yubF. (i) Plasmid Construction. Nucleotide sequence
pGJHy2k, therefore contained the yubF gene inactivated by
from the B. subtilis complete genome sequence (26) was used
the insertion of a kanamycin cassette. Multiple mutant strains
to clone an internal fragment of the lytG gene into pMUTIN4
were made by transformation with the appropriate chromo-
(27) to create plasmid pGJH100. Using genomic DNA as a
template a 300 bp fragment was amplified by PCR using
(ii) Transformation of E. coli and B. subtilis. Transforma-
the forward primer (5’-GCCGAAGCTT64CCGCTAGCTC-
tion of E. coli was performed as described by Hanahan (30). TGTTTGTTT83) and the reverse primer (5’-CGCGGATCC368
Transformation of B. subtilis 168 with pGJH100 and
CGAATGGTTTTTCTTTTCC349), where the restriction sites
pGJHy2k was performed by the competent cell method (31).
are underlined and the internal sequence of the gene is
Confirmation of lytG gene disruption by means of Campbell-
italicized (numbering is with respect to the A of the
type recombination was achieved by Southern blot analysis,
translational start codon of the gene). Nucleotide sequence
using pMUTIN4 HpaI digested fragment containing the
from the B. subtilis complete genome sequence (26) was used
multiple cloning site as a probe. Southern blot analysis, using
to amplify, by PCR, two fragments either side of the yubF
the yubF containing 2.25 kbp fragment from pGJH170 as a
gene and overlapping. One stretching from 982 bp before
probe, showed that the kanamycin resistance cassette had
the start of, to 50 bp into the yubF gene was amplified using
been inserted into the yubF gene by a double crossover event
the forward primer (5’-CGCGCGGATCCCGAAGGAAG-
creating the strain GJH145. Hybridization, probe labeling,
GTT-958) and the reverse primer (5’-TAAGCTAGTACTCTC-
and detection were done using the Boehringer Mannheim
GAGCAACCGTGTTTCTGCGTC13). The other fragment
nonradioactive DNA labeling and detection kit.
stretches from 50 bp in, to 1002 bp after the end of, the
Analysis of Gene Expression. To measure gene expression
gene and was amplified using the forward primer (5’-
during sporulation, synchronous sporulation was performed
GACGCAGAAACACGGTTGCCTCGAGAGTACTAGCT-
using the resuspension method of Sterlini & Mandelstam
TA50) and the reverse primer (5’-GCGAGCTCGCCGACA-
(32). After the initiation of sporulation (t0), samples were
ATCGGCGG1002). The two fragments were placed in a PCR
harvested every hour for 9 h and sporulation morphology
reaction with the two external primers to amplify the whole
monitored by microscopy. Levels of -galactosidase activity
region. The product was cleaned by gel extraction and
were measured as described by Horsburgh and Moir (33).
digested with BamHI and SacI alongside pUC119. The
Cell Separation. The measurement of filamentation and
digested products were ligated by the method of Sambrook
macrofilamentation was performed as described by Blackman
et al. (28). The resultant plasmid pGJH170 was digested with
et al. (13), with the exception of shaking at 40 rpm instead
XhoI that cuts the ligated fragment at the engineered site
of 45 rpm for liquid cultures grown at 25 °C. Biochemistry, Vol. 42, No. 2, 2003 259
Swarm Plate Assay. Swarming motility of strains was
measured on nutrient agar plates (0.3% w/v) as describedby Blackman et al. (13). Cell Autolysis and Preparation of Cell-Wall-BindingProtein (CWBP) Extracts. Lysis of cells of B. subtilis parentaland mutant was performed and cell wall binding proteinsprepared as described by Blackman et al., (13). OVerexpression and Purification of LytG Protein. Nucle-
otide sequence from the B. subtilis complete genomesequence (26) was used to amplify the lytG gene with bothits putative signal sequence and stop codon removed. The801 bp fragment of lytG was amplified using the forwardprimer (5’-GTTTCCATGGCAACTTTATCAAAACCGAT-TGA110) and the reverse primer (5’-TAAACTCGAGGGT-TGCCTCCTTTATTTCA827). The fragment was cleaned bygel extraction and restricted with XhoI and NcoI alongsidepET24d. The restricted products were ligated using themethod of Sambrook et al. (28), creating the plasmidpETGJH6. Transformation of E. coli BL21(DE3) withpGJH6 was as described by Hanahan (30). The cloned lytGgene was sequenced which determined that no errors in itssequence had occurred during amplification (results notshown). Cultures of BL21(DE3) containing pETGJH6 in LB
+ glucose (0.5% w/v) + Kanamycin (30 µg mL-1) weregrown overnight (25 °C, 250 r.p.m) and inoculated 1 in 20into LB with glucose and kanamycin. Cells were grown at25 °C, 250 rpm until OD600 reached 0.6-0.8 at which point(IPTG) was added to a final concentration of 0.4 mM. Samples were taken before the addition of IPTG and athourly intervals for 4 h for SDS-PAGE and renaturingSDS-PAGE analysis. Cells were harvested at 4 h and
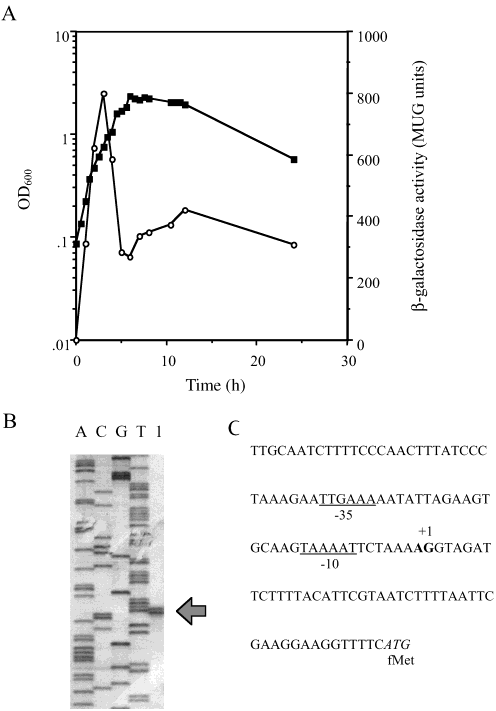
FIGURE 1: The lytG gene is vegetatively expressed. (A) Expression
sonicated, and His-tagged LytG protein was purified using
of lytG::lacZ was measured in GJH100 during growth in NB at 37
HisTrap metal chelate affinity chromatography as described
°C. OD600 (closed symbols), -galactosidase activity (open sym-
in the manufacturers instructions (Amersham Pharmacia
bols). (B) Analysis of the 5′ end of the lytG transcript by primerextension. The sequencing ladder is in lanes A, C, G, and T. Lane
1 is the primer extension of RNA from strain 1A304. (C)
SDS-PAGE and Renaturing SDS-PAGE. Protein samples
Chromosomal DNA sequence proximal to the lytG transcriptional
from overexpression studies and CWBP samples were
start site. The transcriptional and translational starts are shown in
analyzed by 12% (w/v) SDS-PAGE (34) and Coomassie
bold and italics, respectively. The putative -10 and -35 regions
blue staining to visualize proteins. Renaturing gel electro-
phoresis using purified B. subtilis vegetative cell walls as
intervals. Some samples were boiled and treated with
substrate was used to detect autolysin activity as described
Cellosyl as described by Atrih et al. (36). Before purification,
samples were reduced using borohydride, which converts the
Analysis of LytG ActiVity. All assays were carried out at
reducing sugar to its corresponding alcohol. Muropeptides
37 °C in a Victor2 (Wallac), in 96 well plates with B. subtilis
were analyzed by mass spectrometry (MS) measurements
cell walls (0.19 mg mL-1) and purified LytG (20 µg mL-1).
using MALDI-TOF (36). NMR analysis was carried out on
Activity was measured as loss of OD450, with 1 unit of
Bruker DRX-500 and DRX-600 spectrometers. Samples were
enzyme activity being defined as that, which will result in
analyzed using two-dimensional correlated spectroscopy
loss of 0.001 OD450 units/min-1. To determine the optimum
(COSY), total correlated spectroscopy (TOCSY), and rotating
pH, buffer and cation for LytG activity sodium citrate,
frame nuclear Overhauser effect spectroscopy (ROESY). For
phosphate, and Tris/HCl buffers with pH’s ranging from 3
TOCSY, a spin-lock field of 9 kHz was used over a mixing
to 6.3, 5.8 to 8.0, and 7 to 9, respectively were used. The
time of 95 ms, while for ROESY, a spin-lock field of 3 kHz
presence of 20mM of the cations, MgCl2, CaCl2, CuCl2,
was used over a mixing time of 150 ms. Spectra were
HgCl2, or ZnCl2, the chelator EDTA, or a range of MgCl2
processed using FELIX (Accelrys Inc., San Diego, CA).
and CaCl2 concentrations (0-100mM) were also tested. Analysis of lytG Promoters. Primer extension was carried
out as described previously (34). Analysis of Enzymatic Hydrolysis of Peptidoglycan by RP-Analysis of lytG Expression. Expression of lytG was
HPLC. Preparation of cell wall peptidoglycan and separation
measured using a lacZ reporter fusion in strain GJH100.
of soluble muropeptides were carried out as previously
Maximal expression was observed during the mid-exponen-
described (35) but with the following modifications. Pepti-
tial phase of vegetative growth (Figure 1A), with none during
doglycan (2 mg mL-1) was hydrolyzed with purified LytG
sporulation (results not shown). Total RNA was purified from
(50-200 µg mL-1) at 37 °C with samples taken at defined
the wild-type strain at the estimated time of maximal
260 Biochemistry, Vol. 42, No. 2, 2003
FIGURE 2: Role of autolysins in cell separation. Formation of boliin liquid cultures occurs due to the increased filamentation in strainGJH110 (lytC lytD lytG) mutant (B) compared to strain SH128(lytC lytD) mutant (A). Liquid cultures (10 mL nutrient broth) weregrown overnight at 25 °C with gentle shaking (40 rpm) and pouredinto Petri dishes for photography.
expression (1.5-2 h) of lytG. The transcriptional site for lytGwas determined by primer extension to be located 48/49 bpupstream of the lytG translational start site (Figure 1B). Thepromoter has a -35 and -10 region of TTGAAA andTAAAAT, respectively. Role of LytG. Cell Growth and DiVision. In nutrient broth
(37 °C, 250 rpm) all B. subtilis (Table 1) strains grew atequivalent rates and reached the same final OD600 (resultsnot shown). Although accurate readings could not be obtainedfor strains SH131(lytC lytD sigD) and GJH110 (lytC lytDlytG) due to hyperfilamentation of cells. When strains weregrown at 25 °C with shaking at 40 rpm clumps of cells (boli)became visible in the medium in strain GJH110 (lytC lytD
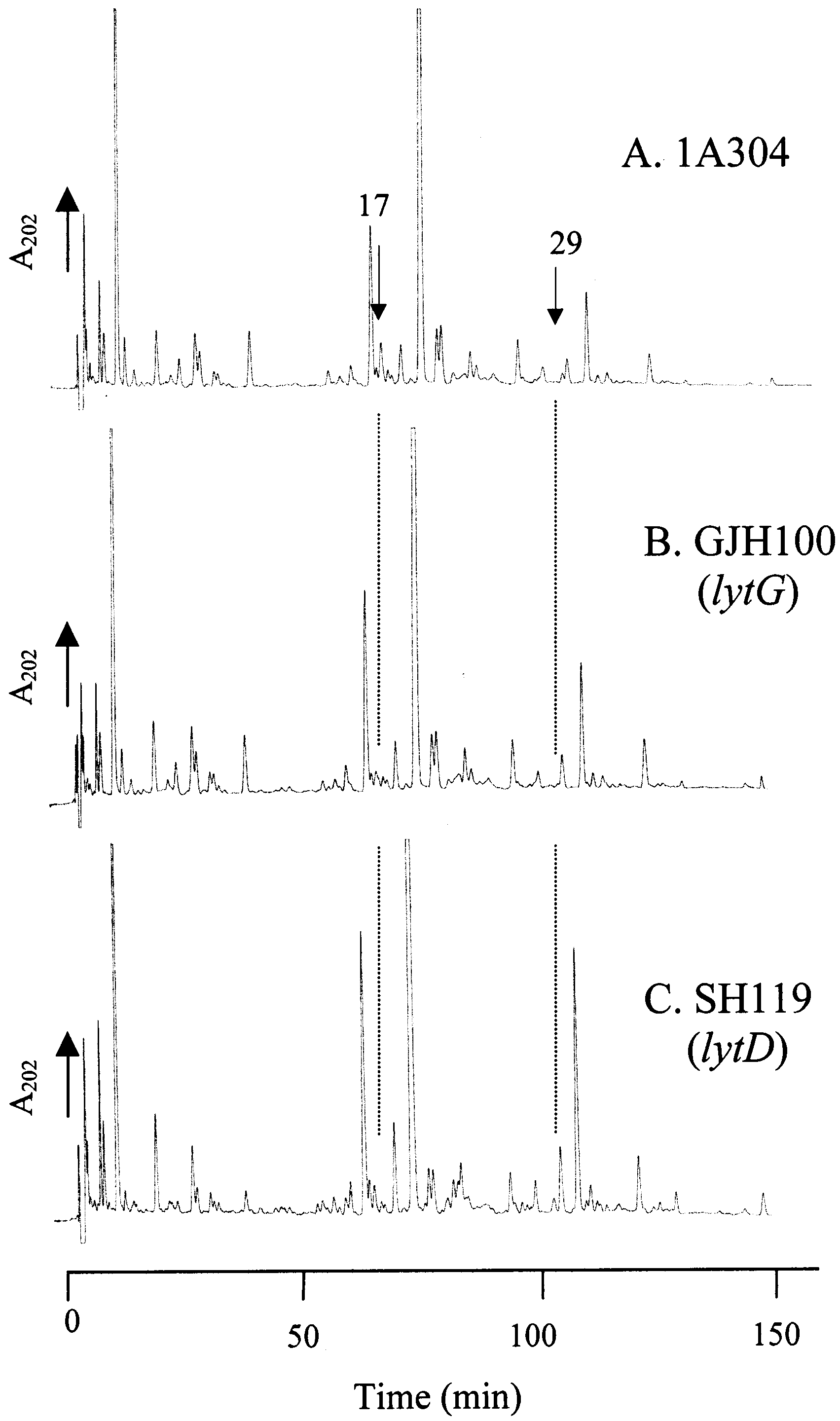
FIGURE 3: Role of LytD and LytG in B. subtilis peptidoglycanstructural determination. RP-HPLC muropeptide elution patterns
lytG) but not in strain SH128 (lytC lytD) (Figure 2).
of peptidoglycan from B. subtilis 168 vegetative cells of 1A304
However, SH115 (lytC), SH119 (lytD), and GJH100 (lytG)
(wild type) (A), GJH100 (lytG) (B), and SH119 (lytD) (C).
showed no such degree of boli formation. After growth at25 °C, 40 rpm, strains GJH110 (lytC lytD lytG) and SH128(lytC lytD) were both observed to be highly filamentous by
was exacerbated (16% lysis after 90 min treatment). A similar
compensatory role for LytD and LytG in lysis in response
Swarming Motility. Strains SH115 (lytC),GJH100 (lytG),
to sodium azide was also noted (results not shown).
and GJH104 (lytC lytG) were motile in stationary phase when
Role of LytG in in ViVo Peptidoglycan Structural Deter-
viewed by phase-contrast microscopy. These strains were
mination. B. subtilis has a characteristic muropeptide profile
analyzed on swarm plates, which measures chemotaxis as
during vegetative growth (Figure 3A) (35). Analysis by RP-
well as bacterial motility. Motile strains form a halo of diffuse
HPLC of purified cell walls of wild type, strain SH119 (lytD),
growth round a tightly packed central colony on swarm
and strain GJH100 (lytG) showed a substantial reduction in
plates. Strain SH115 (lytC) showed a reduction in swarming
muropeptide 17 and a complete loss of muropeptide 29 (see
motility compared to its parent 1A304, the halo diameter
Figure 3; muropeptide designation from ref 35). Muropeptide
being 44 (SD ( 0.7) versus 74 (SD ( 0.5) mm. This result
17 was previously shown to contain two products named
is consistent with the findings of Blackman et al. (13). Strain
17a and 17b, corresponding to disaccharide tripeptide dis-
GJH100 (lytG) also showed a reduction in swarming motility,
accharide tetrapeptide with 1 phosphate and two amidations
the halo diameter being 48 (SD ( 1.3) mm. GJH104 (lytC
and disaccharide tripeptide disaccharide tetrapeptide with two
lytG) demonstrated an even greater reduction in swarming
amidations and missing a glucosamine (35). Muropeptide
(32 (SD ( 0.4) mm). No swarming motility defect was noted
29 was also previously identified as disaccharide tripeptide
for strain GJH145 (yubF). SH128 (lytC lytD) and GJH110
disaccharide tetrapeptide disaccharide tetrapeptide missing
(lytC lytD lytG) do not swarm at all (as previously noted for
a glucosamine (35). In contrast SH119 (lytD) (Figure 3C)
showed no alteration in muropeptide profile compared to
Cell Autolysis. Inactivation of lytG (strain GJH100) alone
had no significant effect on lysis by cloxacillin (results not
Analysis of Cell-Wall-Binding Protein Extracts. Renaturing
shown). Also, in combination with LytC, there was no defect
SDS-PAGE analysis of cell-wall-binding protein (CWBP)
over and above LytC. However, although SH119 (lytD) only
extracts of wild type and mutant strains revealed that LytG
showed a modest reduction in lysis rate in the presence of
was not a major CWBP and that its action could not be
cloxacillin compared to 1A304 (79% vs 90% lysis after 90
demonstrated on zymograms from whole cell extracts (results
min treatment, respectively), in GJH144 (lytG lytD) the effect
Biochemistry, Vol. 42, No. 2, 2003 261
FIGURE 4: LytG has glucosaminidase activity. RP-HPLC muropep-tide elution patterns of soluble peptidoglycan fragments from B. subtilis 168 (HR) vegetative cells hydrolyzed with: (A) Cellosyl,(B) LytG, or (C) LytG and Cellosyl. Role of LytF. GJH145 (yubF) had no phenotypic differ-
ences from the wild type (1A304) in any of the above assays(results not shown). Analysis of LytG ActiVity. LytG protein was overexpressed
in E. coli and purified by virtue of a C-terminal His tag. The purified protein showed maximal activity in sodiumcitrate buffer (10 mM, pH 5.7). Activity was greatest in the
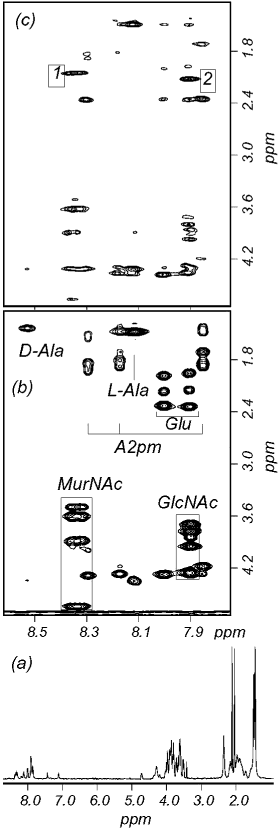
FIGURE 5: 500 MHz NMR spectra of muropeptide 5(1). Spectra
were acquired in water containing 10% D2O at 35 °C. (a) 1D
spectrum. The water resonance was suppressed by presaturation
EDTA (results not shown). The inhibition of LytG activity
and has been removed by a low-frequency digital convolution filter.
by EDTA suggests that LytG was purified with chelated
(b) Part of the 2D TOCSY spectrum, indicating spin system
Mg2+ and that the presence of that cation is essential for
assignments. The A2pm signals correspond to correlations with both
LytG activity. Under optimal conditions hydrolysis (as
the R and δ amides. Correlations involving the sugar resonances
measured by loss of optical density) was observed with
are boxed. (c) Part of the 2D ROESY spectrum, same spectral regionas part (b). The cross-peaks marked 1 and 2 are discussed in the
purified M. luteus and B. subtilis cell walls but not with S.aureus, S. mutans, E. faecalis, or L. arabinosus (results notshown). The optimal specific activity of the purified enzyme
ever, the muropeptide elution pattern from HPLC shows that
was 450 U mg-1 using B. subtilis vegetative cell walls as
these muropeptides have different retention times than their
equivalents observed by Atrih et al. (35) after muramidase
Determination of LytG Hydrolytic Bond Specificity. B.
(Cellosyl) digestion. This difference could be accounted for
subtilis 1A304 purified peptidoglycan was hydrolyzed with
by the relative position of glucosamine residues. To deter-
purified LytG under conditions which resulted in <5% lossof OD
mine the structures of these muropeptides, NMR analysis
450 and the soluble material was analyzed (Figure 4B).
Two major peaks were observed (peaks 4 and 5, Figure 4B)
was performed. One-dimensional spectra were typical of
which did not correspond to any of the previously character-
muropeptides. In particular, the spectrum of muropeptide 5(1)
ized major Cellosyl derived muropeptides (Figure 4A). The
(Figure 5a) contained two signals between 7.0 and 7.5 ppm,
novel products were collected, desalted, and analyzed by
with intensities corresponding to single protons, indicating
mass spectroscopy. On desalting it was observed that peak
that 5(1) has a single amidation on one of the two available
5 consisted of two muropeptides. Both of these (called
A2pm residues. By contrast, the spectrum of 5(2) (data not
muropeptides 5 (1) and 5 (2)) were collected and analyzed
shown) indicates two amidations, one on each A2pm residue.
by mass spectroscopy and amino acid analysis (Table 2).
The spectrum of muropeptide 5(1) (Figure 5a) has two
This identified these three muropeptides as disaccharide
prominent intense peaks at 2.04 and 2.11 ppm. We have
tripeptide (muropeptide 4) and disaccharide tripeptide dis-
previously shown (35) that these can be assigned to the acetyl
accharide tetrapeptide (muropeptides 5(1) and 5(2)). How-
methyls of MurNAc and GlcNAc, respectively, and do not
262 Biochemistry, Vol. 42, No. 2, 2003
change position significantly on reduction of the reducing
not sufficiently diagnostic to allow us to say with confidence
which resonances come from MurNAc and which from
The structures of the muropeptides were determined using
GlcNAc. This assignment is made most unambiguously from
2D COSY, TOCSY, and rotating frame NOE (ROESY)
the ROESY spectrum (Figure 5c) which contains intense
spectra, and the method is illustrated using spectra of
cross-peaks (labeled 1 and 2), respectively from the MurNAc
muropeptide 5(1) (Figure 5). The spin systems are clearly
and GlcNAc 2′-amide proton to its adjacent acetyl methyl.
identifiable from TOCSY spectra (Figure 5b), which indicate
This demonstrates that the reducing end is GlcNAc. Other
for example two closely similar sets of sugar resonances
sequential NOEs confirm the expected disaccharide tripeptide
coupled to 2′-amide protons at around 8.35 ppm (left-hand
disaccharide tetrapeptide structure.
box in Figure 5b), and two more sets coupled to amide
After partial hydrolysis of B. subtilis 1A304 peptidoglycan
protons at around 7.9 ppm (right-hand box in Figure 5b).
with LytG as above, total material was further hydrolyzed
The chemical shifts indicate that the sets around 8.35 ppm
by Cellosyl, which resulted in >95% loss of OD450, and the
retain characteristic pyranose sugar frequencies (in particular,
soluble products were analyzed by HPLC. Two apparently
the anomeric proton at around 4.7 ppm), while the sets
novel muropeptides (2 and 3) were observed in the LytG
around 7.9 ppm are from the reduced alcohol, i.e., this sugar
and Cellosyl digested material (Figure 4C) compared with
is the reducing end. However, the chemical shift values are
the Cellosyl control (Figure 4A), and a large increase in peak
Biochemistry, Vol. 42, No. 2, 2003 263
1 was also noted (Figure 4C). Muropeptides 1, 2, and 3
change, or steric hindrance, due to the presence of two
(Figure 4C) were collected, desalted, and analyzed by mass
amidations, may prevent the action of Cellosyl on both
spectroscopy and amino acid analysis (Table 2). Muropep-
potential cleavage sites in 5(2). Thus, the amidation state
tides 1, 2, and 3 (Figure 4C) are muramic acid tripeptide,
effects peptidoglycan hydrolase activity. This may form a
monosaccharide tripeptide monosaccharide tetrapeptide, and
novel mode of posttranslational regulation of peptidoglycan
disaccharide tripeptide disaccharide tetrapeptide missing one
hydrolase activity via this subtle modification of the stem
glucosamine, respectively (Table 2). For muropeptide 3 the
monosaccharide may be substituted with either the tri- or
Hydrolysis of peptidoglycan with LytG alone resulted in
the appearance of only disaccharide containing muropeptides
A digestion time course also failed to reveal any muropep-
(Figure 4B). These were monomer and dimer muropeptides.
tides other than disaccharides (muropeptides 4 and 5). Also
All three contained only disaccharide substitutions. Larger
Cellosyl digestion of soluble LytG products did not give any
muropeptides were never recovered in significant quantities.
products apart from muropeptides 1, 2, and 3 (results not
Thus LytG is, at least primarily, an exoenzyme acting
processively from the ends of the glycan strands. This is thefirst autolytic exo-glucosaminidase to be described in B.DISCUSSION subtilis 168. The enzyme was able to efficiently hydrolyzeonly cell wall peptidoglycan from B. subtilis and M. luteus
Sequence homology had suggested that LytG may be a
but not S. aureus and other species, which may be due to
muramidase (25). Thus, it was surprising that lack of LytG
different substrate chemical structures. O-Acetylation of
resulted in the disappearance of muropeptides from the B.
muramic residues in S. aureus may be responsible for this
subtilis 168 peptidoglycan profile that corresponded to those
lack of activity, which has also been shown to prevent the
which had been previously predicted to be due to glu-
hydrolysis of S. aureus peptidoglycan by lysozyme (38).
cosaminidase activity, as a result of the lack of an N-
Also, HF treatment of B. subtilis cell walls led to an increase
acetylglucosamine residue at the nonreducing terminus (35).
in peptidoglycan hydrolysis rate by LytG. Teichoic acid and
In contrast, missing the major, well characterized glu-
membrane phospholipids have previously been proposed to
cosaminidase, LytD, did not affect the muropeptide profile.
have moderating effects on LytD activity (8).
Thus, it was proposed that LytG may be a glucosaminidase
LytG is a new member of the family of vegetative
and constitutes the major activity of this type involved in
autolysins of B. subtilis (25). These enzymes perform, in
peptidoglycan hydrolysis in the intact cell, even though it is
many cases, overlapping and mutually compensatory roles
not a major cell wall protein. The lack of LytD products
in multiple physiological functions. A number of autolysins
may be due to the absence of appropriate substrate for this
have been shown to be involved in separation of daughter
enzyme and stringent posttranslational control of activity.
cells after septation (6, 39). LytC, LytD, and LytG are all
Despite intense speculation the biochemical regulation of
involved in boli formation (probably due to hyperfilamen-
autolysins in situ which prevents spontaneous autolysis has
tation), as only the triple mutant forms such large clumps.
remained elusive. Very recently it has been shown that the
Such levels of functional redundancy may be the result of
cell wall of B. subtilis is protonated during growth (37). Both
the importance of cell separation in the natural environment
LytD and LytG have slightly acid pH optima, which suggests
and the presence of the autolysins in the exposed location
they may well be active in the cell wall environment.
of the cell wall open to attack by proteases etc. LytG also
To verify the hydrolytic bond specificity of LytG, the
plays a role in motility and chemotaxis, as evidenced by a
protein was overexpressed and purified. Muropeptide analysis
defect in swarming ability. This could be due to slight
of LytG hydrolyzed peptidoglycan revealed unequivocally
changes in filamentation status, which would impact on
that LytG is a glucosaminidase (Table 2 and Figure 4).
chemotactic ability, or flagellar extrusion may be effected
Cellosyl (muramidase) digestion of LytG hydrolyzed pep-
by aberrant cell wall hydrolysis. Lysis due to azide or cell
tidoglycan results in the loss of muropeptides 4 and 5 (Figure
wall antibiotics occurs as a post-mortem event. In this case
4B) and the appearance of 1, 2, and 3 (Figure 4C). Thus
LytG and LytD showed mutually compensatory roles.
muropeptides 4 and 5 are glucosaminidase-derived substrates
Transcriptional fusion and primer extension analysis
for the muramidase. Muropeptide 4 is hydrolyzed by Cellosyl
revealed lytG transcription to occur from a single promoter,
to produce muropeptide 1. Muropeptides 5(1) and 5(2) are
similar to the consensus sequence typical of σA-dependent
muramidase hydrolyzed to make muropeptides 2 and 3,
promoters (40). This is in contrast to lytC and lytD, which
respectively, as revealed by their relative amidation status
are completely or in part under the control of the alternative
(Table 2). This confirms that LytG is a novel glucosamini-
sigma factor σD. Thus, LytG may have its primary function
dase unrelated to previous enzymes of this class. The
during exponential growth. The roles of the large complement
homology of LytG to AcmA suggests that either related
of seemingly compensatory autolysins during growth and the
enzymes may have different activities or that previous
mechanisms whereby all these components combine to create
methodologies used to determine hydrolytic bond specificity
a functional cell wall architecture have remained obscure.
have not been stringent enough. Thus, it is important to verify
The classical representation of peptidoglycan is as long
activity using the sensitive RP-HPLC assay.
glycan strands cross-linked periodically by peptide side
The presence of muropeptide 3 (which retains an N-
chains. However evidence from Escherichia coli (41) and
acetylglucosamine residue) suggests that Cellosyl is unable
recently from S. aureus (42) have revealed this convention
to fully hydrolyze muropeptide 5(2). Muropeptide 2 was
to be misleading. The average glycan strand length in S.
shown to have only one amidation by NMR, while muropep-
aureus is in fact 6 disaccharides. Thus, the bulk of the
tide 3 had two amidations (Table 2). A conformational
peptidoglycan is made up of short chains, which must be
264 Biochemistry, Vol. 42, No. 2, 2003
highly cross-linked in order to maintain integrity. The
18. Brewer, S. J., and Berkeley, R. C. W. (1973) Biochem. J. 134,
presence of oligomer muropeptides linking multiple chains
19. Ortiz, J. M., Berkeley, R. C. W., and Brewer, S. J. (1973) J. Gen.
alludes to a specific organization within the peptidoglycan.
The accurate chain length of peptidoglycan of B. subtilis has
20. Ortiz, J. M. (1974) J. Bacteriol. 117, 909-910.
not yet been determined. The activity of glucosaminidases,
21. Foster, S. J. (1992) J. Bacteriol. 174, 464-470.
such as LytG and LytD, will be to modify glycan strand
22. Margot, P., Wahlen, M., Gholamhuseinian, A., Piggot, P., and
Karamata, D. (1998) J. Bacteriol. 180, 749-752.
length. Their role in this crucial parameter for peptidoglycan
23. Ishikawa, S., Hara, Y., Ohnishi, R., and Sekiguchi, J. (1998) J.
structure is currently under investigation.
24. Margot, P., Pagni, M., and Karamata, D. (1999) Microbiology 145,
REFERENCES
25. Smith, T. J., Blackman, S. A., and Foster, S. J. (2000) Microbiology
1. Ghuysen, J.-M., Tipper, D. J., and Strominger, J. L. (1966)
26. Kunst, F., Ogasawara, N., Moszer, I., and 148 other authors (1997)
2. Rogers, H. J., Thurman, P. F., and Burdett, I. D. J. (1983) J Gen.
27. Vagner, V., Dervyn, E., and Ehrlich, S. D. (1998) Microbiology
3. Fein, J. E., and Rogers, H. J. (1976) J. Bacteriol. 127, 1427-
28. Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989) Molecular
4. Fein, J. E. (1979) J. Bacteriol. 137, 933-946. Cloning: a Laboratory Manual, 2nd ed., Cold Spring Harbor
5. Pooley, H. M., and Karamata, D. (1984) J. Bacteriol. 160, 1123-
29. Gue´rout-Fleury, A.-M., Shazand, K., Frandsen, N., and Stragier,
6. Ward, J. B., and Williamson, R. (1984) in Microbial Cell WallSynthesis and Autolysis (Nombela, C., Ed.) pp 159-166, Elsevier,
30. Hanahan, D. (1983) Mol. Biol. 166, 557-580.
31. Anagnostopoulos, C, and Spizizen, J. (1961) J. Bacteriol. 81, 741-
7. Herbold, D. R., and Glaser, L. (1975) J. Biol. Chem. 250, 1676-
32. Sterlini, J. M., and Mandelstam, J. (1969) Biochem. J. 113, 29-
8. Rogers, H. J., Taylor, C., Rayter, S., and Ward, J. B. (1984) J.Gen. Microbiol. 130, 2395-2402.
33. Laemmli, U. K. (1970) Nature 227, 680-685.
9. Kuroda, A., and Sekiguchi J. (1991) J. Bacteriol. 173, 7304-
34. Horsburgh, M. J., and Moir, A. (1999) Mol. Microbiol. 32, 41-
10. Lazarevic, V., Margot, P., Soldo, B., and Karamata, D. (1992) J.
35. Atrih, A., Bacher, G., Allmaier, G., Williamson, M. P., and Foster,
Gen. Microbiol. 138, 1949-1961.
S. J. (1999) J. Bacteriol. 181, 3956-3966.
11. Margot, P., and Karamata, D. (1992) Mol. Gen. Genet. 232, 359-
36. Atrih, A., Zo¨llner, P., Allmaier, G., Williamson, M. P., and Foster,
S. J. (1998) J Bacteriol. 180, 4603-4612.
12. Margot, P., Maue¨l, C., and Karamata, D. (1994) Mol. Microbiol.
37. Calamati, H. G., Ehringer, W. D., Koch, A. L., and Doyle, R. J.
(2001) Proc. Natl. Acad. Sci U.S.A. 98, 15260-15263.
13. Blackman, S. A., Smith, T. J., and Foster, S. J. (1998) Microbiol.
38. Snowden, M. A., Perkins, H. R., Wyke, A. W., Hayes, M. V.,
and Ward, J. B. (1989) Microbiology 135, 3015-3022.
14. Helmann, J. D., Marquez, L. M., and Chamberlin, M. J. (1988) J.
39. Forsberg, C., and Rogers, H. J. (1971) Nature 229, 272-273.
40. Haldenwang, W. G. (1995) Microbiol. ReV. 59, 1-30.
15. Rashid, M. H. Mori, M., & Sekiguchi, J. (1995) Microbiology
41. Harz, H., Burgdorf, K., and Holtje, J.-V. (1990) Anal. Biochem.
16. Ortiz, J. M., Gillespie, J. B., and Berkeley, R. C. W. (1972)
42. Boneca, I. G., Huang, Z.-H., Gage, D., and Tomasz, A. (2000) J.Biochim. Biophys. Acta 289, 174-186.
17. Berkeley, R. C. W., Brewer, S. J., Ortiz, J. M., and Gillespie, J.
B. (1973) Biochim. Biophys. Acta 309, 157-168.
Acta Orthop. Belg. , 2005, 71 , 29-35 Anastomosis between the median and ulnar nerve in the forearm An anatomic study and literature review Konstantin J. KAZAKOS, Anastasios SMYRNIS, Konstantin C. XARCHAS, Alexandra DIMITRAKOPOULOU, From the Orthopaedic Department, Democritus University of Thrace, Alexandroupolis, Greece Anastomosis between the median and ulnar nerve in and finally
PLACINGS FOR BOLDMERE SC 11 CLUB CHAMPIONSHIPS at Stechford Female swimmers 6 points for 1st place, 5 points for 2nd place and so on: swimmers with equal points are listed in alphabetical order. AGE GROUP A AGE GROUP B AGE GROUP C 1st Irisha POWELL (BLDM) 1st Erin DAVIES (BLDM) 1st Harriet GORDON (BLDM) (04) (A) 24 points (0
 Biochemistry, Vol. 42, No. 2, 2003 259
Swarm Plate Assay. Swarming motility of strains was
measured on nutrient agar plates (0.3% w/v) as describedby Blackman et al. (13).
Biochemistry, Vol. 42, No. 2, 2003 259
Swarm Plate Assay. Swarming motility of strains was
measured on nutrient agar plates (0.3% w/v) as describedby Blackman et al. (13).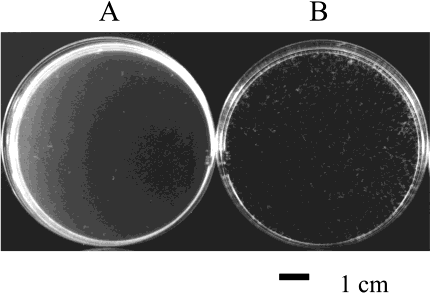
 260 Biochemistry, Vol. 42, No. 2, 2003
FIGURE 2: Role of autolysins in cell separation. Formation of boliin liquid cultures occurs due to the increased filamentation in strainGJH110 (lytC lytD lytG) mutant (B) compared to strain SH128(lytC lytD) mutant (A). Liquid cultures (10 mL nutrient broth) weregrown overnight at 25 °C with gentle shaking (40 rpm) and pouredinto Petri dishes for photography.
260 Biochemistry, Vol. 42, No. 2, 2003
FIGURE 2: Role of autolysins in cell separation. Formation of boliin liquid cultures occurs due to the increased filamentation in strainGJH110 (lytC lytD lytG) mutant (B) compared to strain SH128(lytC lytD) mutant (A). Liquid cultures (10 mL nutrient broth) weregrown overnight at 25 °C with gentle shaking (40 rpm) and pouredinto Petri dishes for photography.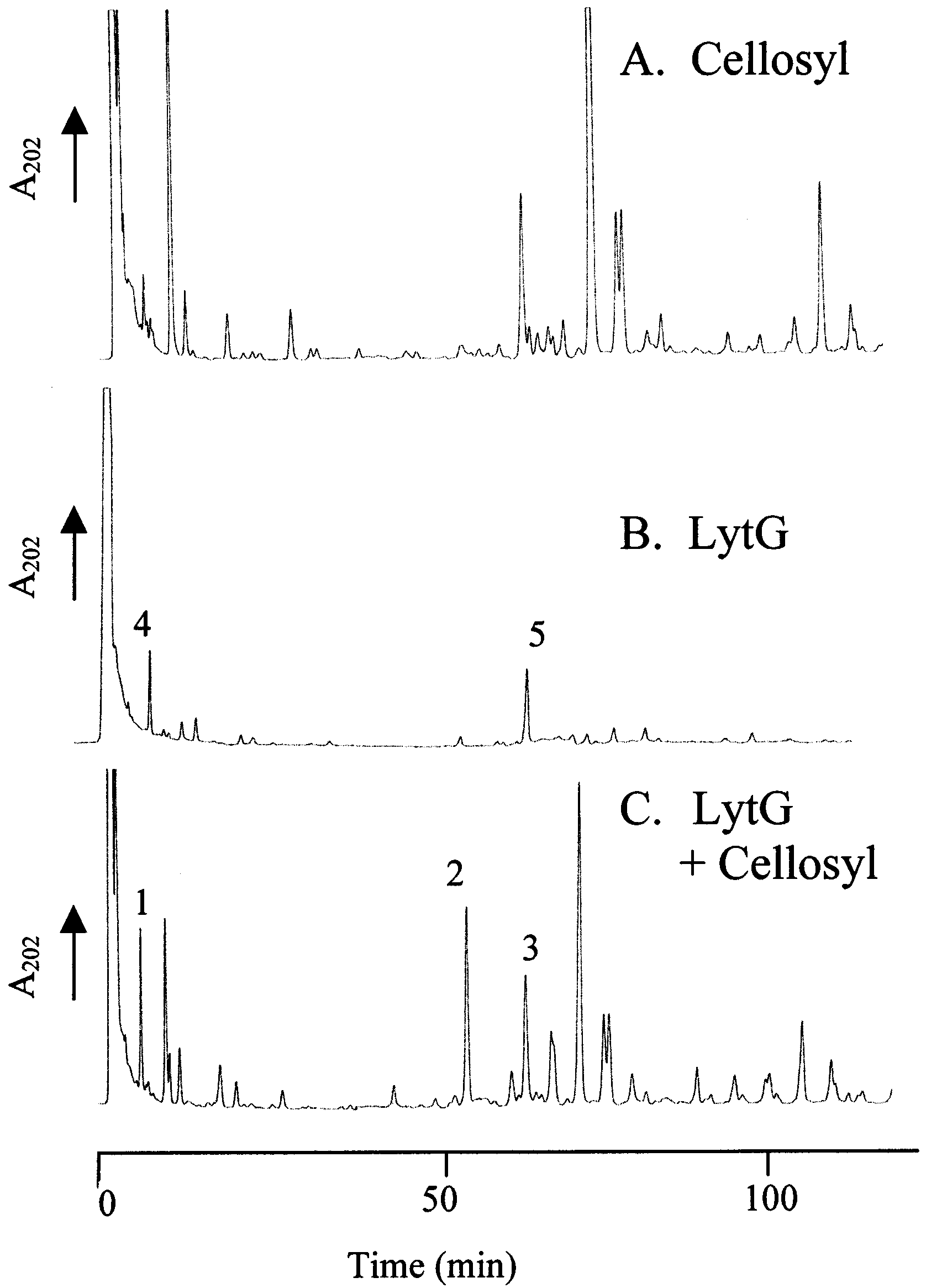
 Biochemistry, Vol. 42, No. 2, 2003 261
FIGURE 4: LytG has glucosaminidase activity. RP-HPLC muropep-tide elution patterns of soluble peptidoglycan fragments from B.
Biochemistry, Vol. 42, No. 2, 2003 261
FIGURE 4: LytG has glucosaminidase activity. RP-HPLC muropep-tide elution patterns of soluble peptidoglycan fragments from B.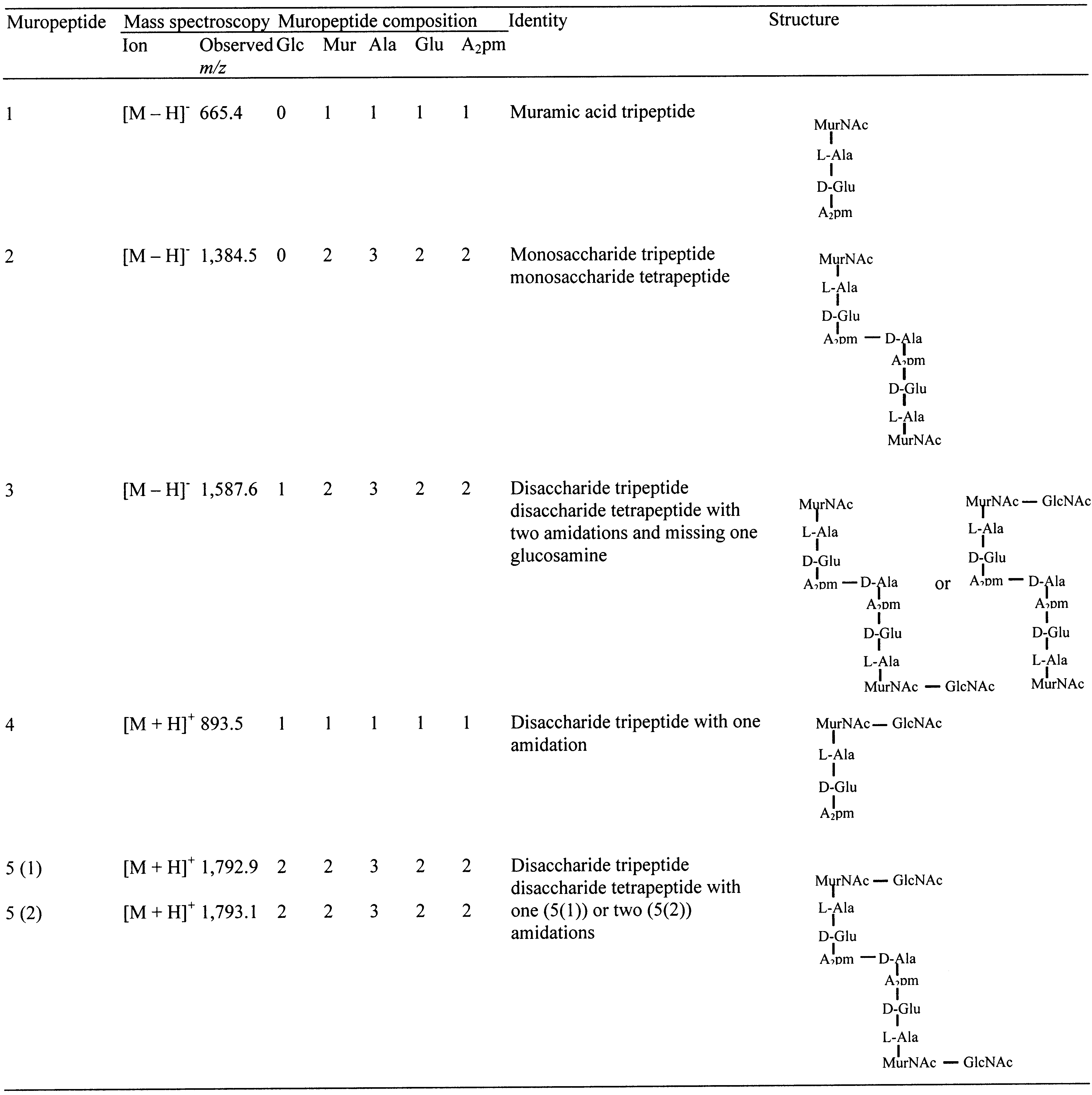 262 Biochemistry, Vol. 42, No. 2, 2003
change position significantly on reduction of the reducing
not sufficiently diagnostic to allow us to say with confidence
which resonances come from MurNAc and which from
The structures of the muropeptides were determined using
GlcNAc. This assignment is made most unambiguously from
2D COSY, TOCSY, and rotating frame NOE (ROESY)
the ROESY spectrum (Figure 5c) which contains intense
spectra, and the method is illustrated using spectra of
cross-peaks (labeled 1 and 2), respectively from the MurNAc
muropeptide 5(1) (Figure 5). The spin systems are clearly
and GlcNAc 2′-amide proton to its adjacent acetyl methyl.
262 Biochemistry, Vol. 42, No. 2, 2003
change position significantly on reduction of the reducing
not sufficiently diagnostic to allow us to say with confidence
which resonances come from MurNAc and which from
The structures of the muropeptides were determined using
GlcNAc. This assignment is made most unambiguously from
2D COSY, TOCSY, and rotating frame NOE (ROESY)
the ROESY spectrum (Figure 5c) which contains intense
spectra, and the method is illustrated using spectra of
cross-peaks (labeled 1 and 2), respectively from the MurNAc
muropeptide 5(1) (Figure 5). The spin systems are clearly
and GlcNAc 2′-amide proton to its adjacent acetyl methyl.