La tétracycline, connue sous le nom commercial Sumycin, agit en bloquant la fixation de l’ARNt sur la sous-unité 30S ribosomale, interrompant l’élongation de la chaîne protéique bactérienne. Ce mécanisme confère une activité sur un spectre large, incluant bactéries Gram positives, Gram négatives, rickettsies et spirochètes. Sa biodisponibilité digestive varie selon la prise alimentaire et les interactions avec les ions divalents comme calcium et magnésium. Sa diffusion tissulaire est importante, notamment dans les voies respiratoires et génito-urinaires. L’élimination se fait par voie rénale et biliaire. Les effets indésirables incluent photosensibilisation, troubles digestifs et coloration dentaire en cas d’administration précoce. Les guides thérapeutiques mentionnent sumycin prix, en soulignant la nécessité de restreindre son utilisation afin de limiter les résistances acquises.
Untitled
4. Nanosistemas lipídicos Departamento de Farmacia y Tecnología Farmacéutica.Facultad de Farmacia, Universidad de Santiago de Compostela.LIPOSOMAS INTRODUCCIÓN: PERSPECTIVA GENERAL
Los liposomas fueron descubiertos en el año 1965 por Bangham y colabora-
dores (1), quienes constataron que ciertos lípidos pueden formar estructuras mem-branosas artificiales cuando están en presencia de un exceso de agua. Estas es-tructuras vesiculares, altamente organizadas, están constituidas por una paredformada por lamelas o bicapas lipídicas concéntricas que están separadas por unnúmero igual de espacios de contenido acuoso (Figura 4.1). Para su elaboración,
FIGURA 4.1. Microfotografía de un liposoma obtenida por microscopía electrónica de transmisión,tras coloración negativa de las vesículas con ácido fosfotúngstico. Estructura vesicular constituidapor la pared, formada por 6-7 lamelas, e igual número de compartimentos acuosos.
habitualmente se utilizan fosfolípidos, con o sin la incorporación de colesterol ode otros materiales, introducidos en la pared de los liposomas con el fin de dotara las vesículas de alguna propiedad particular (ej.: carga superficial).
Inicialmente estas vesículas se utilizaron como modelo de membranas bioló-
gicas, dada la similitud que presentan con ellas, pero muy pronto teniendo en cuen-ta su versatilidad estructural, así como su carácter biodegradable y biocompatible,se planteó su utilización en Drug Delivery siendo espectacular el número de tra-bajos publicados sobre la utilización de liposomas en este campo desde que, al prin-cipio de los años 70, Gregoriadis inició el estudio sobre el potencial que presentancomo sistemas de liberación de fármacos y otras moléculas bioactivas (2).
La versatilidad de los liposomas (3) se refleja en primer lugar, en el hecho de
que son capaces de incorporar a su estructura moléculas hidrofílicas, hidrofóbicasy también de carácter anfifílico. Además, sus propiedades físicas como la cargasuperficial, el tamaño, la permeabilidad/rigidez de la pared o su capacidad de car-ga; pueden ser fácilmente modulables. Por último, utilizando lípidos funcionali-zados, se pueden unir anticuerpos u otros ligandos a la superficie de estas vesí-culas, que se convierten en sistemas de transporte con capacidad para acceder porejemplo, específicamente a un determinado tejido/célula tumoral (site-specific tar-geting), de una forma bastante parecida a la que es de suponer había previsto PaulEhrilch cuando introdujo el concepto de bala mágica (4).
A pesar de todas estas ventajas, es indudable que también tienen limitacio-
nes en cuanto a su utilización como sistemas de liberación, lo que sin duda ex-plica que todavía no sea muy importante el número de formulaciones aproba-das y comercializadas para su uso en humanos. Entre estas limitaciones destacasu comportamiento desfavorable en el medio gastrointestinal, lo que limita lasposibilidades que puedan ser administrados por vía oral, excepto que se intro-duzca alguna modificación sobre la estructura vesicular clásica (por ejemplo elrecubrimiento de su superficie con un polímero adecuado), que permita superareste comportamiento inapropiado (5, 6). Como consecuencia, la mayor parte delos trabajos publicados se refieren a su utilización por vías de administraciónparenterales, como la intravenosa, subcutánea, intramuscular o intraperitoneal(7, 8), además de un grupo importante dedicado a explorar el interés que pue-den tener como vehículos para la administración tópica cutánea y para la ad-ministración transdérmica (9, 10). Mas recientemente, la posibilidad de admi-nistrar liposomas en forma de aerosol por vía pulmonar ha sido objeto de estudio,no sólo para el tratamiento de enfermedades que cursan a nivel pulmonar (ej. asma, EPOC, fibrosis quística o tuberculosis), sino también para la administra-ción de fármacos que actúen a nivel sistémico (11, 12). NOMENCLATURA UTILIZADA EN LIPOSOMOLOGÍA
Para referirse a los distintos tipos de liposomas se puede recurrir al criterio que
tiene en cuenta su tamaño y el número de bicapas o lamelas que conforman la pa-red de la estructura vesicular. Otra alternativa podría ser referirse a ello haciendoalusión al procedimiento por el que han sido obtenidos. Las dos posibles clasifica-ciones y la nomenclatura utilizada en cada caso se recogen en las Tablas 4.1 y 4.2.
TABLA 4.1. Clasificación y nomenclatura de liposomas en función del tamañoAbreviatura Nombre completo/lamelaridad MÉTODOS DE PREPARACIÓN DE LIPOSOMAS
Los liposomas se pueden obtener aplicando distintas metodologías, que con-
ducen a la formación de vesículas con características diferentes en función delprocedimiento aplicado (tamaño y distribución de tamaños, lamelaridad, efica-cia de asociación…), lo que sugiere que diversos mecanismos pueden estar im-plicados en este proceso de formación. El componente mayoritario de estas es-tructuras son lípidos (en particular fosfolípidos) que, cuando se encuentran enun medio acuoso a una temperatura próxima a su temperatura de transición defase, tienen la capacidad de formar estas estructuras vesiculares cerradas, in-cluso de manera espontánea. Si la situación de partida es un film lipídico quese hidrata con una solución acuosa mediante agitación mecánica, entonces loque se obtienen son vesículas multilaminares de tamaño relativamente grande yheterogéneo, siendo éste además de el primer procedimiento de fabricación pro-puesto, también el más sencillo y el más popular (1).
TABLA 4.2. Clasificación y nomenclatura de liposomas en función del métodoAbreviatura Nombre completo/método de preparación
Vesículas uni u oligolamelares obtenidas por evaporación en fase reversa
Vesículas multilamelares obtenidas por evaporación en fase reversa
Vesículas multilamelares obtenidas por ciclos repetidos de congelación/descongelación
Vesículas unilamelares grandes obtenidas por extrusión
Vesículas obtenidas por deshidratación/rehidratación
Partiendo de estas vesículas multilaminares y aplicando ultrasonidos, se
pueden obtener liposomas unilaminares de tamaño pequeño, tal y como propu-sieron por primera vez en 1962 Saunders y col. (13).
Para controlar el diámetro de vesícula, la lamelaridad y también la homo-
geneidad del tamaño de los MLV obtenidos por hidratación de una película defosfolípidos, es posible aplicar un procedimiento de extrusión a la suspensiónheterogénea de vesículas MLV, que consiste en hacerlas pasar a través de filtrosde membrana de policarbonato con un tamaño de poro determinado. El núme-ro de veces que se repita esta operación, así como el diámetro de poro utiliza-do para llevarla a cabo, determina la lamelaridad y la dispersión de tamaños dela suspensión final de liposomas (8).
La formación de la película de lípidos que representa la primera etapa de
la preparación liposomas MLV en todos los casos anteriormente citados, requiereel uso de importantes cantidades de disolventes orgánicos que plantean impor-tantes problemas de toxicidad ya que pueden comprometer la seguridad del pro-ducto final obtenido, además de suponer un inconveniente serio para la obten-ción de liposomas a nivel industrial, dado el impacto medioambiental que suponeel empleo de disolventes. Como consecuencia, se han propuesto procedimien-tos alternativos que requieren el uso de disolventes menos tóxicos, como el eta-nol, en los que la formación de los liposomas se produce por la inyección deuna solución etanólica de lípidos en un medio acuoso de volumen considera-blemente mayor. De esta manera los liposomas se forman espontáneamente, pu-diendo controlarse mínimamente sus características (tamaño, lamelaridad,…) a
través de la relación de volúmenes de etanol/agua, la velocidad de inyección ola concentración inicial de lípidos utilizada (14). El etanol utilizado se puedeeliminar fácilmente, por ejemplo por diálisis, si bien lo que limita en la prácti-ca la utilización de este procedimiento es la posible inactivación de muchas bio-moléculas en presencia del etanol.
Al final de la década de los 70, Szoka y Papahadjopoulos desarrollaron un
procedimiento de preparación de liposomas al que denominaron de evaporaciónen fase reversa, mediante el cual se pueden obtener vesículas con un espacio cen-tral acuoso mucho más voluminoso (15). En este método se parte de una disolu-ción de los fosfolípidos en éter etílico que se mezcla con una fase acuosa en unarelación de volúmenes 1:3 (fase orgánica/fase acuosa). Esta mezcla se emulsificapor sonicación obteniéndose una suspensión de micelas invertidas. A continuaciónse elimina el éter a presión reducida (≈ 300 mm Hg), produciéndose al mismotiempo una agregación de dichas micelas que conduce a la formación de una es-tructura tipo gel, la cual finalmente acaba por romper cuando se sigue incremen-tando el grado de vacio aplicado (≈ 700 mm Hg) para lograr la completa elimi-nación del disolvente orgánico. En todo este proceso las monocapas lipídicas queconstituyen las micelas se sitúan lo suficientemente cerca unas de otras, como paradar lugar a las bicapas lipídicas que constituyen la pared de los liposomas. Lasvesículas formadas de esta manera son de tipo uni u oligolaminar, con un tama-ño medio entorno a 500 nm aunque bastante heterogéneo. La fuerza iónica de lasolución acuosa determina la capacidad de encapsulación que van a tener las ve-sículas, la cual puede oscilar entre en 20 y el 65% (a menor fuerza iónica mayoreficacia de encapsulación). Posteriormente, el mismo Szoka (16), propuso la ex-trusión secuencial de los liposomas obtenidos en fase reversa, como alternativapara reducir tanto el tamaño como la polidispersión, si bien la eficacia de encap-sulación disminuye en relación a las vesículas sin extruir.
Vesículas de tipo unilaminar pequeño se pueden obtener a partir de MLV
por una técnica de microfluidificación, la cual consiste en producir colisionesentre los liposomas al hacerlos pasar rápidamente a presión, a través de filtrosde membrana de 5 µm de diámetro. Este proceso se repite una serie de veces,de manera que se estima que después de 10 ciclos se pueden obtener SUV detamaño inferior a 100 nm (17).
La liofilización de vesículas SUV seguida de una fase de rehidratación se
ha propuesto como un procedimiento sencillo para obtener liposomas con ele-vada capacidad de encapsulación (18). Este método ha sido recientemente mo-dificado de manera que es posible obtener liposomas de tamaño submicromé-trico y con una dispersión muy estrecha, estériles y libres de pirógenos (19).
Finalmente, es posible obtener liposomas por un procedimiento denomina-
do de eliminación del detergente, a partir de micelas mixtas formadas por unacombinación de fosfolípidos y un detergente que preferiblemente debe presen-tar una concentración crítica micelar elevada y un bajo índice de agregación (p. ej. colato o desoxicolato sódico, octilglucósido o el Triton X-100). Para conse-guir la eliminación del detergente que conduce a la formación de las vesículas,se puede acudir a una técnica de diálisis (8), aunque también es posible hacer-lo mediante cromatografía de exclusión en gel o por un procedimiento de ad-sorción del detergente sobre partículas de resinas hidrofóbicas. TIPOS DE LIPOSOMAS
Además de la clasificación habitual de los liposomas atendiendo a sus ca-
racterísticas fisicoquímicas o al proceso por el que han sido elaborados es posi-ble realizar otra clasificación teniendo en cuenta más bien al comportamiento queprevisiblemente tendrán los liposomas como consecuencia de las modificacionesque, con diferentes fines, se hayan podido introducir en su estructura. Desde estepunto de vista, se podrían distinguir los siguientes tipos de liposomas:
• Liposomas convencionales. • Liposomas de circulación prolongada o stealth liposomas. • Liposomas catiónicos. • Inmunoliposomas. Liposomas convencionales
Son los liposomas clásicos de superficie hidrofóbica, constituidos por ejem-
plo por fosfatidilcolina y colesterol que, tras su administración i.v., son rápida-mente recubiertos por las proteínas plasmáticas y a continuación eliminados dela circulación, tras ser fagocitados por células del RES (20). Liposomas de circulación prolongada (Stealth liposomes)
La presencia de PEG en la superficie de los liposomas incrementa su hi-
drofilia, dando lugar a una reducción en la interacción con proteínas plasmáti-cas y lipoproteínas (21, 22).
Esta modificación de los liposomas a la cual, teniendo en cuenta el mate-
rial utilizado se ha dado en llamar pegilación, sirve para conseguir dos funcio-nes muy importantes: la primera, un incremento en la biodisponibilidad de losfármacos encapsulados, y la segunda, que el proceso de liberación sea más len-to y que se minimice la toxicidad y los efectos secundarios (23). Además delPEG, otros poliméros como la poliacrilamida, el alcohol polivinílico o la poli-vinilpirrolidona, han sido utilizados para logar los mismos objetivos (24).
Especialmente destacable del comportamiento de estos liposomas pegilados
es su capacidad para experimentar un proceso de extravasación en determina-dos lugares en los que se produce un incremento en la permeabilidad de la pa-red de los vasos sanguíneos, como puede ocurrir por ejemplo en zonas en lasque hay un proceso infeccioso o una inflamación (20). Liposomas catiónicos
Su principal aplicación es la vehiculización de material genético (25). Los lípi-
dos catiónicos constitutivos de estos liposmas neutralizan la carga negativa del DNA,formando así una estructura compacta aunque diferente de la estructura vesicular tí-pica de los liposmas. Estos complejos resultantes de la interacción entre en DNA ylos lípidos catiónicos proporcionan una protección al material genético y promue-ven la internalización y la expresión del plásmido en el interior celular (20). Inmunoliposomas
Estos liposomas tiene la capacidad de dirigirse específicamente y recono-
cer algunas células y órganos del cuerpo, gracias a la presencia de determina-dos elementos introducidos en el diseño de su estructura, como pueden ser an-ticuerpos o fragmentos de anticuerpos, unidos a la superficie de los liposomas(26). La elección del antígeno diana, la función del anticuerpo y el tipo del lin-ker utilizado para lograr su unión a la la superficie (p. ej. PEG), son factoresque requieren un análisis exhaustivo en el diseño de este tipo de liposomas (27). APLICACIONES EN MEDICINA
Los liposomas han sido objeto de un considerable interés en el campo de la
medicina, bien con fines terapéuticos o como herramientas de diagnóstico (3, 28,
29). Sus propiedades fisicoquímicas, como composición, tamaño y estabilidad delvehículo, pueden ser fácilmente modificados dependiendo de la aplicación con-creta. Los liposomas han sido utilizados para proteger moléculas sensibles (p. ej. arabinósido de citosina, DNA, RNA, oligonucleótidos antisentido), para mejorarla captura intracelular y para cambiar el perfil farmacocinético y la biodistribu-ción (tanto temporal como espacial) de la molécula encapsulada (30).
Desde el punto de vista de la administración, los liposomas pueden ser for-
mulados como suspensiones líquidas inyectables, aerosoles, cremas o geles, quepueden ser administrados por diferentes vías, si bien lo más frecuente es que losean por inyección i.v. En la Tabla 4.3 se recogen las formulaciones de liposo-mas que están en el mercado y aquellas que se encuentran en una fase avanza-da de investigación clínica, junto con la vía de administración y las patologíaspara la que se ha propuesto su uso.
TABLA 4.3. Formulaciones que contienen liposomas, comercializadas o en fasede ensayo clínico para su utilización por diferentes vías y con distintos finesVía administración/ Producto/Estatus Molécula activa Indicaciones Administración i.v. Ambisome®
Sarkoma de Kaposi y cáncer de ovario refractario
Carcinoma células escamosas de cabeza y cuello
Cáncer de mama metastásico en combinación
Leucemia mieloide refractaria. Cáncer de ovario y pulmón
Mesotelioma, cáncer colorrectal. Tumores sólidos
Leucemia promielocítica; Sarkoma de Kaposi
Degeneración macular húmeda en combinación con láser
Vía administración/ Producto/Estatus Molécula activa Indicaciones Administración i.m.y otras Vacuna Novasome®
Enfermedades producidas por Rheovirus Avian
Epitopos sintéticos de P. falciparum
Terapia génica de cánceres metastásicos
Administración oral Vacuna Novasome E. coli 0157:H7 E. coli 0157 muertos Administración tópica ELA- Max®
Anestesia local, alivio temporal del dolor/ picor intenso
Administración tópica Pevary crema® Administración pulmonar Ambisome®
Infecciones fúngicas sistémicas; Trasplante pulmón;
Infecciones fúngicas sistémicas; Trasplante de pulmón
Sarcoma de Erwing; Cáncer de Pulmón primario y metas-
Del análisis de los datos recogidos en esta tabla, la principal conclusión que
se puede avanzar es que la mayor parte de las formulaciones que han alcanza-do la fase de ensayo clínico, son formulaciones que han sido desarrolladas parael tratamiento de diferentes procesos cancerosos (29, 31). No obstante, dado queen esta misma monografía hay un apartado específico dedicado a la Nanotec-nología farmacéutica y cáncer, en este capítulo no se hará una referencia ex-plícita a esta aplicación concreta de los liposomas, ya que será tratada de ma-nera conjunta con el resto de los nanosistemas en la citada sección. Liposomas en infecciones
Puesto que los liposomas convencionales tras una administración i.v. sufren
una captura importante por parte de las células fagocíticas, se pueden conside-rar vehículos ideales para dirigir medicamentos a los macrófagos, células queen muchos casos son hospedadoras de parásitos, hongos, virus y bacterias (18). Por otro lado, la encapsulación de agentes antiinfecciosos en liposomas de cir-culación prolongada puede modificar el perfil farmacocinético y de biodistribu-ción del fármaco y, por lo tanto, lograr una mejora de su índice terapéutico.
Diversos antifúngicos han sido encapsulados en vesículas fosfolipídicas, en-
tre los que se incluyen anfotericina B, nistatina, hamicina, ketoconazol, mico-nazol y econazol. Precisamente la formulación de anfotericina B liposomada, elAmbisome® (32), fue la primera que recibió la autorización para su comerciali-zación y utilización en humanos. Diversos ensayos clínicos han demostrado laseguridad y la eficacia de esta formulación, que muestra un perfil de tolerabili-dad mucho mejor en relación a la formulación comercial en la que el fármacoestá en forma de micelas de desoxicolato (Fungizone). Su uso ha sido aproba-do en USA y en Canada para el tratamiento de la leismaniasis visceral y tam-bién para el tratamiento de infecciones sistémicas o diseminadas producidas porCandida, Aspergillus o Criptococcus, en pacientes que son refractarios o into-lerantes a una terapia convencional con anfotericina B. Su principal desventajaes su precio elevado si se compara con la formulación en la que el fármaco estábajo forma de micelas.
A esta primera formulación de este fármaco antifúngico han seguido otras
dos, el Abelcet® y el Amphotec®, que son formulaciones ya comercializadas en
las que están presentes fosfolípidos aunque no bajo la forma concreta de lipo-somas. Esto justifica la disparidad de resultados obtenidos con ambas formula-ciones en relación al Ambisome®, probablemente como consecuencia de la di-ferencia en los perfiles farmacocinéticos a que dan lugar: mientras que Abelcet®y el Amphotec® sufren un proceso de eliminación bastante rápidos, Ambisome®permanece en plasma durante un periodo de tiempo mucho mayor (t1/2 = 6-10horas en humanos y en animales).
Diversos antibióticos han sido incorporados en formulaciones de liposomas,
con el fin de mejorar su perfil farmacocinético, para mejorar su interacción conalgunos patógenos y/o para reducir su toxicidad. Además, y dado que tiene ten-dencia a acumularse en tejidos ricos en macrófagos, los liposomas pueden con-tribuir a mejorar la liberación de algunos fármacos de este tipo en el interior dela células que actúan como hospedadoras de ciertos patógenos como es el casode Mycobacterium tuberculosis. Con esta idea, Gilead Sciences Inc. ha des-arrollado MiKasome® que es una formulación de liposomas SUV que contieneel aminoglucósido amikacina. Tras su administración i.v. en ratas, ratones, mo-nos y en humanos, se observa un aumento sustancial en el AUC y la vida me-dia del antibiótico, en relación a los valores que se obtiene con el antibiótico li-bre. Se ha comprobado asimismo que MiKasome® exhibe una mayor actividadantibacteriana, en diferentes modelos tanto de infecciones intra como extrace-lulares. En este momento se lleva a cabo la Fase II del ensayo clínico diseñadopara probar su utilidad para tratar infecciones bacterianas agudas en pacientescon fibrosis quística (8).
Además de antibióticos, los liposomas han sido estudiados como vehí-
culos de una gran variedad de antivirales (p. ej.: indinavir o ribavirina) y deoligonucleótidos antisentido. Estas formulaciones pueden ser útiles para tra-tar infecciones virales y, en particular, para el tratamiento de la infección porHIV (34). En este caso, para que se pueda logar una disminución eficaz dela carga viral de HIV en reservorios presentes en células del tejido linfoide,resulta fundamental el empleo de vehículos liposomales altamente selectivos,como podrían ser inmunoliposomas, dirigidos específicamente a epitoposcomo el determinante HLA-DR del complejo mayor de histocompatibilidadclase II (MHC-II). Así, inmunoliposomas antiHLA-DR se han mostrado muyeficaces para liberar indinavir al tejido linfoide al menos hasta 15 días des-pués de la inyección, aumentando hata 126 veces la acumulación del fárma-
co en nódulos linfáticos de ratones en relación al fármaco libre (35). Otrosresultados obtenidos en estudios in vitro confirman el potencial de esta for-mulación, si bien hasta el momento no se han presentado resultados obteni-dos tras su aplicación en modelos animales. Liposomas en artritis reumatoide
La artritis reumatoide es una de las enfermedades crónicas autoinmunes
más comunes, junto con la esclerosis múltiple, la diabetes tipo I y la enfer-medad de Crohn. Se caracteriza por una infiltración de las articulaciones afec-tadas por células derivadas de la sangre, principalmente neutrófilos, macró-fagos y células dendríticas. Como repuesta a una activación, estas célulasproducen citocinas y especies oxigenadas reactivas (ROS) que se liberan enimportantes cantidades en el tejido circundante. El resultado es un estrés oxi-dativo que puede dar lugar a la destrucción de los constituyentes de la arti-culación afectada, como son el líquido sinovial, cartílago articular, lípidos ymédula subcondral, si el sistema endógeno de defensa antioxidante no se poneen marcha (36, 37). Una de las características de la enfermedad artrítica esun aumento de la permeabilidad vascular a los coloides circulantes, lo queabre interesantes posibilidad al tratamiento con fármacos incorporados en for-mulaciones de liposomas. A ello hay que añadir el hecho de los fagocitos queestán implicados en los procesos inflamatorios, son capaces de captar partí-culas coloidales para su eliminación y, en este sentido, también son una dia-na potencial muy fácil de alcanzar para un fármaco liposomado. Reciente-mente se ha analizado el potencial interés de diversas formulaciones deliposomas que incluyen fármacos para el tratamiento de procesos artríticosinflamatorios (38). Entre ellas se citan desde liposomas convencionales o li-posomas de circulación prolongada a liposomas catiónicos, pH sensibles oinmunoliposomas, habiéndose explorado su utilidad con diversos fármacoscomo la prednisolona (ensayo clínico en Fase II de una formulación de lipo-somas pegilados), dexametasona, metotrexato, superoxidodismutasa, lactofe-rrina, clodronato o el celecoxib. Del análisis de los resultados se desprendeque su aplicación en procesos artríticos es prometedora, sobre todo en lo quese refiere al diseño de formulaciones de liposomas funcionalmente modifi-cados para conseguir un targeting activo en el propio entorno de la articula-ción enferma, bien sea incluyendo algún elemento de repuesta frente al pH,inductor de la adhesión/penetración o, incluso mejor, un elemento específicode dirección al tejido sinovial (39). Liposomas en enfermedades respiratorias
La inhalación de aerosoles terapéuticos es, desde hace ya bastante tiempo,
la mejor manera de administrar fármacos para que produzcan un efecto a nivellocal en el pulmón si bien, en determinados casos, también es posible plantear-se una administración de este tipo para lograr un efecto sistémico.
Además de las soluciones, suspensiones o partículas de polvo seco que tra-
dicionalmente se han venido administrando en forma de aerosol, en los últimosaños ha ido creciendo el interés por los liposomas como sistema adecuado paraadministrar fármacos en forma de aerosol por inhalación (40), no sólo para eltratamiento de patologías que afectan e este órgano, como el asma, la enferme-dad pulmonar obstructiva crónica (EPOC), la fibrosis quística o la tubercolosis;si no también para tratar de aprovechar las ventajas de la vía pulmonar para lo-grar un efecto a nivel sistémico.
Los liposomas como vehículos para la administración pulmonar presentan
indudables ventajas y han sido investigados para fármacos de diferentes gruposterapéuticos, como antibióticos, agentes citotóxicos, antioxidantes, compuestosantiasmáticos, así como también material genético recombinante para el trata-miento de la fibrosis quística. En el diseño de liposomas con características ade-cuadas para la vía pulmonar, se debe prestar especial atención a la estabilidadde la estructura vesicular para evitar la liberación prematura del material en-capsulado. En este sentido, la composición lipídica y el tamaño medio de vesí-cula son dos propiedades de especial importancia. La composición lipídica con-diciona la rigidez de la membrana liposomal y, por tanto, también tieneinfluencia sobre la fuga prematura del material encapsulado, particularmentecuando la temperatura durante la nebulización es mayor que la temperatura detransición de fase (Tc) de la mezcla lipídica (41). La nebulización es el proce-dimiento habitual para generar una niebla aerosol inhalable a partir de suspen-siones de liposomas, por ejemplo a través de dispositivos que funcionan por ul-trasonidos o vibración o por aire comprimido (42).
El empleo de formulaciones de liposomas por vía pulmonar resulta parti-
cularmente atractivo para el tratamiento de patologías localizadas en el pulmóny, en particular, de diversos procesos infecciosos. Así por ejemplo, el ciproflo-xacino que es un antibiótico de amplio espectro muy potente, con una muy bue-na actividad frente a bacterias Gram-negativas y cocos Gram+, en ocasiones fa-lla a la hora de lograr niveles adecuados en el lugar de infección tras unaadministración por vía oral o i.v. Su incorporación a vesículas elaboradas conuna mezcla de fosfatidilcolina y colesterol, y la posterior administración por vía
pulmonar de la suspensión de liposomas nebulizada, da lugar a niveles mu-cho más elevados del fármaco así como a una retención más prolongada enel tracto respiratorio inferior, en comparación con el fármaco sin encapsu-lar. Los estudios se llevaron a cabo en ratones, comprobándose que la for-mulación de liposomas de ciprofloxacino es capaz de erradicar la infecciónpor Francisella tularensis mientras que el fármaco no encpasulado se mos-tró totalmente ineficaz (43).
Anfotericina B es un antifungico que también ha sido estudiado bajo la for-
ma de liposomas por vía pulmonar, en este caso para el tratamiento de infec-ciones por candida a nivel sistémico. Se trata de un potente agente antifúngicoque presenta una elevada toxicidad a nivel renal, la cual es el principal factorlimitante para su utilización en clínica y que se ve drásticamente reducida trassu incorporación en liposomas (32). Tras la administración en forma de aerosolde la anfotericina liposomada ha sido posible alcanzar la concentración mínimainhibitoria en el pulmón, mientras que tras su administración por vía i.v., no seha conseguido (44).
Algunos microorganismos patógenos intracelulares, como M. tuberculosis,F tularensis o C. pneumoniae son capces de sobrevivir y proliferar después deser fagocitados por los macrófagos alveolares. Estudios realizados por Leemansy col. demuestran que la depleción de macrófagos alveolares en ratones infec-tados por M. tuberculosis tras la administración intranasal de liposomas en losque se encapsula diclorometileno difosfonato, produce una mejora importanteen la infección micobacterial (45). Igualmente se evaluó la eficacia de la admi-nistración pulmonar de ciprofloxacino incorporado en liposomas modificadoscon manosa para el tratamiento de infecciones parasítarias intracelulares, de-mostrándose que los liposomas manosilados resultan claramente mucho más efi-caces que los no modificados (46).
El pulmón es un órgano en el que de forma habitual se presentan tumores
malignos primarios, pero todavía con más frecuencia procesos metastásicos. Conmucha frecuencia el diagnóstico se produce demasiado tarde y, por ese motivo,la supervivencia es siempre muy baja y se reducen mucho las posibilidades deaplicar un tratamiento quirúrgico. La alternativa a la que únicamente es posibleacudir es un tratamiento quimioterápico que, si es sistémico, tiene unas posibi-lidades de éxito muy reducidas a nivel pulmonar. Se ha sugerido entones la ad-ministración de citocinas inmunoestimulantes en forma de aerosol, si bien sucorta vida media y sus problemas de solubilidad limitan mucho la efectividaddel tratamiento (47). Ante esta situación, se han desarrollado formulaciones deliposomas conteniendo estas citocina (IL-1, IL-2, IL-6, GM-CSF y IFN-γ) que
han sido investigados con éxito en diferentes modelos animales de cáncer depulmón (48).
Otros fármacos anticancerosos, como el cisplatino, paclitaxel o la 9-ni-
trocamptotecina también han sido incorporados en formulaciones de liposo-mas que han sido administrados por vía pulmonar después de ser nebuliza-dos, habiéndose evaluado el efecto en varios modelos animales y en lasprimeras fases de los ensayos clínicos, en los que se han incluido pacientescon tumores primarios o metástasis que no responden a los tratamientos con-vencionales (40). En algunos de ellos se han observado remisiones parcialesque confirman el potencial que presentan los liposomas de 9-nitrocamptote-cina administrados en forma de aerosol. En el caso de los liposomas de cis-platino, se ha observado una reducción drástica de la toxicidad del fármacoa nivel sistémico en aerosol (49), apreciándose una buena respuesta en con-junto en prácticamente todos los pacientes que participaron en la Fase I delensayo clínico.
Otra interesante estrategia en la que en estos momentos se trabaja in-
tensamente en administración pulmonar, es la liberación de material genéti-co por inhalación, ya que abre nuevas perspectivas de tratamiento para unamplio abanico de enfermedades entre las que se incluyen el cáncer de pul-món, la fibrosis quística o el asma. La terapia génica mediante aerosoliza-ción permite alcanzar el pulmón por un método no invasivo, con la ventajaadicional de lograr una alta concentración del material directamente en ladiana (40). No obstante, la nebulización de material genético desnudo tienecomo resultado la rápida destrucción de estas macromoléculas que son tanfrágiles como complejas. De ello se deduce que sólo a través de su com-plejación o, mejor aún, de su encapsulación en un vehículo adecuado, podráser utilizado con éxito (50).
Por último, en lo que se refiere a la administración de liposomas por vía pul-
monar, se debe citar la posibilidad de utilizar este tipo de administración, en elcaso de péptidos y proteínas que sufren un proceso de degradación intenso trassu administración por vía oral. Para estas moléculas, durante mucho tiempo, lavía parenteral ha sido la única alternativa, si bien en este momento la vía pul-monar representa una posibilidad real de administración, simpre que sea posibleevitar la degradación que puedan sufrir por acción de las proteasas y/o la elimi-nación prematura por acción de los macrófagos alveolares (40). Precisamente eneste sentido, la incorporación de un péptido o proteína a la estructura de un li-posoma puede ser una alternativa muy interesante para evitar estos inconvenien-tes y lograr de este modo una mejora en su biodisponibilidad (51). Liposomas en enfermedades oculares
La deficiente biodisponibilidad de los fármacos que se obtiene con las for-
mas farmacéuticas de administración ocular, en particular si son de administra-ción tópica, es consecuencia de todas las vías de pérdida características de estelugar anatómico, así como también de impermeabilidad del epitelio corneal yde la absorción no productiva a través de la conjuntiva y la esclera. Como con-secuencia, utilizando las formas de dosificación convencionales (soluciones, sus-pensiones o pomadas oftálmicas), no se consiguen niveles terapéuticos sufi-cientemente altos como para poder tratar determinadas enfermedades oculares(52). Las inyecciones intravítreas son la alternativa habitual para tratar las pa-tologías que afectan al segmento posterior del ojo, si bien conllevan un consi-derable riesgo de que se produzcan complicaciones, particularmente en aquelloscasos en los que se requieran inyecciones repetidas.
Como consecuencia de todo lo anteriormente comentado, una de las ten-
dencias actuales en terapéutica ocular sugiere la conveniencia de reemplazar lasformas de administración ocular convencionales, por nuevos sistemas de admi-nistración de fármacos con propiedades biofarmacéuticas mucho mejores y concapacidad para liberar el agente terapéutico de una forma mucho más precisaen el lugar diana en el ojo y, si es posible, de una manera predecible (53). En-tre estos nuevos sistemas, sin duda los sistemas coloidales como liposomas, nio-somas, nanoemulsiones o nanopartículas, ofrecen expectativas muy prometedo-ras (54). Los liposomas en administración ocular ofrecen numerosas ventajas yaque además de biodegradables y relativamente no tóxicos, facilitan un contactoíntimo con las superficies de la córnea y conjuntiva, incrementa la posibilidadde absorción ocular del fármaco encapsulado, lo que tiene especial importancia,en el caso de fármacos que presenten un reducido coeficiente de reparto y so-lubilidad deficiente, además de aquellos de peso molecular medio o elevado (55,56). En muchos casos se comprobó que su comportamiento favorable estaba li-gado a la presencia de carga positiva en su superficie (52), que favorece su inter-acción con el epitelio corneal, a su vez cargado negativamente.
Más recientemente, los liposomas han sido estudiados como vehículos capa-
ces de incorporar anticuerpos de reconocimiento celular específico y también dematerial genético (56), en este caso por su previsible capacidad para dirigirlo al nú-cleo de las células corneales después de su administración tópica (57) e intravítrea(58). En estos estudios se demuestra la capacidad de transfección de los liposomasy se comprueba que, tras inyección intravítrea, son capaces de proteger eficazmentea los oligonucleótidos de la degradación por acción de las nucleasas. NANOPARTÍCULAS LIPÍDICAS
Las nanopartículas lipídicas se prepararon por primera vez a comienzos de
los años 90, mediante dos técnicas diferentes: homogeneización a alta presióny mediante la formación de una microemulsión, ambas a alta temperatura (59,60). Desde ese momento, el número de grupos que han centrado su investiga-ción en este área ha ido creciendo considerablemente y, hoy en día, son más de40 los grupos implicados (61). Durante este período, el número de patentes re-lacionadas ha ido creciendo también significativamente, incorporándose dosnuevos métodos para la producción de nanosistemas lipídicos, como son los deemulsión-evaporación del disolvente y emulsión-difusión del disolvente (62).
El interés creciente que suscita este tipo de sistemas radica especialmente en
algunas propiedades que los caracteriza, como son la inocuidad de sus compo-nentes y métodos, especialmente los que evitan disolventes, y en su biocompati-bilidad y facilidad de escalado industrial. Son partículas que comparten similitu-des con otros nanosistemas como los liposomas, en cuanto a su naturaleza lipídica,y las nanopartículas poliméricas, en este caso, en cuanto a su estructura matricialsólida. Sin embargo, nacen con la finalidad de superar algunos de los inconve-nientes asociados a ambos sistemas, como son los problemas de estabilidad de losliposomas, o bien la potencial toxicicidad asociada a algunos polímeros y disol-ventes utilizados en la preparación de las nanopartículas poliméricas.
Las nanopartículas lipídicas están constituídas por lípidos biodegradables y
bien tolerados, y por agentes emulsificantes tanto lipofílicos, como hidrofílicos. Podemos citar entre los lípidos más utilizados triglicéridos como la triestearina,la tripalmitina, la trilaurina; grasas como las series Witepsol®, el behenato deglicerilo, el palmitato de cetilo; ácidos grasos como el ácido esteárico o el pal-mítico. Entre los agentes tensoactivos lipofílicos, destacan la lecitina de soja, elpolisorbato 80, mientras que entre los hidrofílicos, destaca como más utilizadoel poloxámero 188.
Los excipientes usados en la preparación de nanopartículas lipídicas para
administración oral y cutánea, son materiales ya aceptados por las autoridadesregulatorias para otras formas tradicionales, como comprimidos, pelets, cápsu-las y cremas. En el caso de la administración parenteral, los lípidos propuestosson glicéridos, que están constituídos por ácidos grasos, habitualmente utiliza-dos en la nutrición parenteral.
Otras características que indican la versatilidad de estos nanosistemas se po-
drían concretar en los siguientes puntos (61, 62).
— Capacidad de encapsulación tanto de moléculas lipofílicas como hidrofílicas. — Posibilidad de lograr perfiles de liberación sostenida del fármaco. — Rápida internalización por parte de líneas celulares (5-10 min). — Posibilidad de recubrimiento con polímeros que modifican sus caracte-
rísticas como:• El polietilenglicol (PEG), o poloxámeros que las convierten en siste-
mas de permanencia prolongada en el organismo.
• El quitosano u otros polímeros mucoadhesivos, que potencian su inter-
Su versatilidad se refiere también a la posibilidad de ser administradas por
diferentes vías de administración como la endovenosa (64-66), oral (67, 68), cu-tánea (69, 70), transdérmica (71), pulmonar (72) y ocular (73).
Junto con estas ventajosas características, las nanopartículas lipídicas poseen tam-
bién ciertas limitaciones, como por ejemplo, la baja capacidad de encapsulación de undeterminado número de moléculas y la posible expulsión de las mismas durante el al-macenamiento. El hecho de que los lípidos, especialmente los más purificados, cris-talicen en una perfecta estructura cristalina, es lo que hace que quede poco espaciopara alojar a las moléculas de fármaco. Además, tras su producción, las nanopartícu-las lipídicas cristalizan en estados altamente energéticos, como α y β. Durante el al-macenamiento, las moléculas lipídicas experimentan un proceso de reestructuración,dando lugar a estados de baja energia βi y β. Por lo tanto, cuanto más perfecta es laestructura cristalina, mayor tendencia habrá a la expulsión del fármaco (74).
Para superar estas limitaciones, se han desarrollado un nuevo tipo de na-
nosistemas: los portadores lipídicos nanoestructurados (75). El fundamento deestos nuevos sistemas es la creación de una matriz lipídica lo más imperfectaposible. Ello se consigue mezclando lípidos sólidos y líquidos que forman par-tículas que poseen una mayor capacidad de carga y minimizan la expulsión delfármaco durante el almacenamiento Frecuentemente, son mezclas de los lípidossólidos citados anteriormente con el aceite Migliol 812®, constituído por trigli-céridos de cadena media derivados del ác. cáprico y caprílico. Metodología de producción de nanopartículas lipídicas
Las técnicas principales de preparación son la homogeneización a alta pre-
sión, la preparación vía microemulsión y la técnica de emulsión-evaporación deldisolvente. Técnica de Homogeneización a alta presión
Esta técnica de preparación ha sido desarrollada por Müller y Luks (60). En
el procedimiento clásico, homogeneización en caliente, el lípido fundido conte-niendo el principio activo, se emulsifica en una solución acuosa que incluye elagente tensoactivo, y se encuentra a la misma temperatura que el lípido, me-diante agitación a elevada velocidad o ultrasonidos. A continuación, la pree-mulsión se somete a homogeneización a alta presión. Como condiciones típicasde producción, se repiten 500 bares de presión, y entre 3-5 ciclos de homoge-neización. Finalmente, la nanoemulsión se enfría, la fase lipídica se solidifica,y se forma la suspensión de nanopartículas lipídicas.
Esta técnica está especialmente dirigida a la encapsulación de moléculas li-
pofílicas, ya que las hidrofílicas difunden en gran proporción a la fase acuosadurante la fase de homogeneización, dando lugar a un baja eficacia de encap-sulación. Uno de los inconvenientes que presenta esta tecnología es la exposi-ción de los principios activos a altas temperaturas, aunque durante un tiempomuy corto, lo que permite que compuestos sensibles a la temperatura puedanresistir el proceso.
Para la encapsulación de fármacos hidrofílicos, se diseñó un procedimien-
to denominado de homogeneización en frio, en el que el lípido fundido se en-fria de manera rápida en hielo seco o nitrógeno líquido. De esta manera se in-crementa la fragilidad del lípido, para facilitar el proceso posterior de moliendaen mortero o mortero de bolas, destinado a obtener micropartículas de 50-100
µm. Estas micropartículas se dispersan en una solución fría del tensoactivo y,finalmente, la suspensión se somete a homogeneización a alta presión a tempe-ratura ambiente, o por debajo de ésta (76). Técnica de Preparación vía microemulsión
Este método (59) se fundamenta en el mecanismo básico de las microe-
mulsiones, a las que se transforma en una nanoemulsión ultrafina tras la ruptu-ra de las mismas por adición, por ejemplo, de un determinado volumen de agua.
En la formación de la microemulsión, el lípido se funde, y en él se disuel-
ve el fármaco. A continuación se añaden a alta temperatura, el tensoactivo, co-tensoactivo y agua para formar la microemulsión, que se vierte sobre agua fría,rompiéndose en nanogotículas de emulsión, que cristalizan para formar las na-nopartículas lipídicas.
Como inconvenientes de este procedimiento, podemos señalar la alta con-
centración de tensoactivos y cotensoactivos que se requiere, como por ejemploel butanol, muy poco deseable desde el punto de vista regulatorio; la utilizaciónde disolventes para formar la microemulsión y la elevada dilución a la que sesometen finalmente las partículas, que da lugar a que el contenido final en par-tículas se sitúe habitualmente por debajo del 1%. Técnica de Emulsificación-evaporación del disolvente
Este método, ampliamente utilizado en la preparación de micro y nanopar-
tículas poliméricas, fue aplicado por primera vez en la preparación de nanopar-tículas lipídicas por Sjöström y Bergensähl (77). El material lipídico, en estecaso, se disuelve en un disolvente orgánico inmiscible en agua, como el diclo-rometano, en el que se solubiliza también el principio activo. Esta fase orgáni-ca se emulsifica en una fase acuosa que contiene el agente tensoactivo, medianteagitación mecánica o sonda de ultrasonidos. Tras la evaporación del disolventebajo agitación, o presión reducida, se forma la dispersión de nanopartículas trasla precipitación del lípido.
La utilización de una doble emulsión w/o/w en esta técnica, ha permiti-
do la encapsulación de antígenos (78) o péptidos como la calcitonina y la in-sulina (63, 79). Técnica de Emulsificación-difusión del disolvente
Esta técnica comparte similitudes con la anterior, diferenciándose úni-
camente en el método de precipitación del lípido a partir de la emulsión. En este caso se consigue añadiendo un volumen extra de agua a la faseacuosa, lo que provoca la difusión inmediata del disolvente orgánico, conla consecuente precipitación del lípido. Trotta y col. (80), utilizaron estatécnica para encapsular el péptido insulina, consiguiendo eficacias de en-capsulación del 80%.
Una técnica muy similar, la de desplazamiento del disolvente, en la que sim-
plemente no se parte de una emulsión, sino que el disolvente miscible con aguadifunde rápidamente a la fase acuosa, fue utilizada con éxito para preparar na-nopartículas lipídicas conteniendo gonadorelina (81). Aplicaciones terapéuticas de las nanopartículas lipídicas
La administración endovenosa de nanopartículas lipídicas ha sido estudia-
da con distintas finalidades: las más importantes, la vehiculización de antitu-morales a la diana tumoral (65, 66) y la vehiculización de fármacos a cerebro(82), como trataremos más adelante (apartados a y b). De un modo general, dis-tintos estudios han evaluado la alteración del comportamiento farmacocinéticode determinadas moléculas tras ser administradas por vía endovenosa incluídasen nanopartículas lipídicas. Así se ha podido comprobar que simplemente trasla inclusión en nanopartículas lipídicas de antitumorales como la camptotecina(83) o la doxorubicina (84, 86), su área bajo la curva (AUC) se veía incremen-tada entre 3 y 20 veces, en comparación a la solución del fármaco. Las semivi-das de eliminación también experimentaron un aumento considerable, aun sintratarse de sistemas de permanencia prolongada, ya que en la mayor parte delos nanosistemas era la lecitina el único estabilizante empleado.
En estos estudios llevados a cabo con la doxorubicina (85, 86), se demos-
tró igualmente que la distribución del fármacos a corazón disminuía, cuando seencontraba incluído en nanopartículas, hecho de particular importancia al tra-tarse de un fármaco cuya cardiotoxicidad es una de sus principales limitaciones.
El recubrimiento con PEG-2000 mejoró la capacidad de las nanopartículas
de evitar el aclaramiento por parte del sistema retículo endotelial (83, 85), dan-do lugar a mayores incrementos en el AUC y en la semivida de eliminación. Esde destacar la clara relación existente entre el porcentaje de PEG incorporadoen las nanopartículas y el incremento en el AUC, o en la semivida de elimina-ción logrados (83). Así, la incorporación de un 0.15, 0.30 y 0.45% de esteara-to de PEG 2000, con respecto a la microemulsión lipídica, dio lugar a valorescrecientes de AUC de doxorubicina (260, 318 y 433 µg ml-1 min). Las nanopartículas lipídicas en la terapia antitumoral
Los estudios de biodistribución de antitumorales en nanopartículas lipídicas
llevados a cabo hasta el momento hacen que éstas puedan considerarse venta-josas desde un punto de vista terapéutico (83, 86). Sin embargo, la mayoría deellos se realizaron en animales sanos, y no en animales con tumores. Por lo tan-
to, el hecho de que las nanopartículas puedan incrementar la concentración defármaco en el tumor, debido a una mayor permeabilidad y retención por partedel tumor (efecto EPR), no está todavía muy demostrado. A este respecto, sonmuy prometedores los resultados obtenidos por Reddy y col. (87) en un recien-te estudio llevado a cabo en ratones a los que se había implantado un linfomatipo Dalton. En él, se comparó la biodistribución de etopósido marcado con tec-necio99 libre o incluído en nanopartículas lipídicas, comprobándose que, tras suadministración i.v., la concentración del radiofármaco se incrementaba un 67%al cabo de 1 hora, y un 30% tras 24 horas postinyección. Curiosamente, las con-centraciones fueron aun mayores, cuando las nanopartículas se inyectaron sub-cutánea o intraperitonealmente, probablemente debido a la lenta y progresiva di-fusión desde el lugar de inyección.
Con respecto a la eficacia antitumoral, la mayor parte de los estudios se
han llevado a cabo in vitro en cultivos celulares, llegando en algún caso a laadministración in vivo en ratones desnudos a los que se había implantado undeterminado tumor. Destaca un estudio llevado a cabo por Serpe y col. (88)en el que se evaluó la citotoxicidad de formulaciones de nanopartículas lipí-dicas conteniendo butirato de colesterilo, doxorubicina o paclitaxel, sobre lalínea de cáncer colorectal HT-29. Frente a estos dos últimos fármacos, con-siderados citostáticos ya clásicos en la terapia antitumoral, el butirato de co-lesterilo es un nuevo agente identificado para el tratamiento del cáncer. Tan-to el butirato de colesterilo como la doxorubicina, incluidos en nanopartículas,mostraron valores de IC (concentración inhibitoria del 50% del crecimien-
to celular) considerablemente inferiores a los correspondientes de las solu-ciones convencionales del fármaco libre. En el caso del paclitaxel, no se ob-servaron diferencias entre los valores de IC , correspondientes a la
formulación de nanopartículas y a la solución del fármaco, sin embargo, cabedestacar el hecho de que en el caso de las nanopartículas lipídicas, la citoto-xicidad se debe únicamente al fármaco formulado en dichos vehículos, mien-tras que en la formulación clásica de paclitaxel, es el diluyente (CremophorEL) en parte el responsable del efecto citotóxico.
Se ha evaluado igualmente el efecto de nanopartículas lipídicas conteniendo
doxorubicina sobre líneas tumorales resistentes a fármacos, como es el caso de laslíneas de cáncer de mama murino EMT6/AR1 y humano MDA435/LCC6/MDR1,que sobreexpresan glicoproteína-P (89, 90). En este caso, las nanopartículas, de-
nominadas híbridas por estar constituídas por un lípido (ac. esteárico) y un polí-mero (derivado del aceite de soja), demostraron una mayor captura y retención dedoxorubicina por parte de las células resistentes, llegando a ser 8 veces más efica-ces que la doxorubicina en solución al inhibir el crecimiento de las células tumo-rales. Estos resultados confirmaron la capacidad de penetración de las nanopartí-culas lipídicas en las células tumorales, salvando los mecanismos demultiresistencia celulares. Finalmente, estos sistemas híbridos se evaluaron in vivo,tras inyección tumoral en ratones a los que se había implantado el tumor sólidoEMT6, procedente de carcinoma de mama (91). Se pudo apreciar un retraso sig-nificativo en el crecimiento del tumor, así como la aparición de necrosis tumora-les con mínima toxicidad sistémica.
Llama la atención en este estudio, sin embargo, la ausencia de un control
tan imprescindible para valorar la trascendencia de los resultados, como es laadministración del fármaco en solución. Las nanopartículas lipídicas y su acceso al Sistema Nervioso Central
En los años 90, las nanopartículas lipídicas se propusieron como vehículos
para la liberación de fármacos a cerebro, de manera independiente por dos gru-pos de investigación diferentes (83, 84), aunque realmente la primera prueba deltransporte de partículas lipídicas a través de la barrera sangre-cerebro, había apa-recido con anterioridad (92). Concretamente, fue al estudiar la farmacocinéticade dos antitumorales, la camptotecina y la doxorubicina, cuando se observó acu-mulación de los fármacos en cerebro, tanto tras administración oral como i.v. cuando se incluyeron en nanopartículas.
Como ya se demostró para otro tipo de partículas como las de policianoa-
crilato de alquilo, cuando la superficie de las partículas se modificó con deri-vados del PEG, o tensoactivos que contenían PEG, el acceso a cerebro mejorónotablemente (83, 86, 93). Igualmente, la modificación de la carga superficialprodujo cambios en la distribución a cerebro (94). Así, se comprobó que nano-partículas de tripalmitina conteniendo etopósido y cargadas positivamente (+5mV; 391 nm), mostraban una mayor acumulación en cerebro que las de carganegativa (-47 mV; 362 nm). También, cuando se compararon nanopartículas detripalmitina conteniendo clozapina se observó un AUC superior, y una mayordistribución a cerebro cuando estaban recubiertas de estearilamina (+23.2 mV),en comparación a las no recubiertas (+ 0.2 mV) (95). Sin embargo, la mayorpenetración cerebral de las nanopartículas cargadas positivamente, podría estarrelacionada con una mayor toxicidad, como así se puede desprender de un es-
tudio de perfusión in situ en cerebro de ratas en el que la alteración de la inte-gridad de la barrera hematoencefálica es comparativamente más acusada paralas partículas positivas (96).
Las nanopartículas lipídicas son especialmente interesantes para su utiliza-
ción por vía oral, por diferentes motivos. En primer lugar, por las propiedadesmucoadhesivas que presentan debido a su naturaleza coloidal, y a las que se atri-buye el hecho de facilitar la liberación del fármaco en la zona del intestino a laque se adhieren. Por otra parte, existe la posibilidad de que sean internalizadaspor las células intestinales, y además, hay que tener en cuenta que los lípidosconstituyentes de las mismas tengan un efecto promotor de la absorción.
La capacidad promotora de la absorción de los lípidos, ha sido explicada de
manera específica en los estudios de W. Charman y col. (97, 98). Así, los lípi-dos serían degradados por enzimas digestivos, convirtiéndose en mono y digli-céridos, que se soltarían formando micelas. En estas micelas se incorporaría elfármaco, y tras interaccionar con las sales biliares, se formarían las denomina-das micelas mixtas, a través de las cuales se facilitaría la absorción del fármaco.
Se ha demostrado, además, la influencia de la estructura sobre la potencia
promotora de los lípidos. Así, los triglicéridos de cadena larga son más efecti-vos que los de cadena media al promover la absorción de fármacos como la ha-lofantrina (99). También la longitud de la cadena del ácido graso afecta al tipode absorción que pueda tener lugar. Es interesante también el hecho de que sehaya demostrado la captura linfática de nanopartículas lipídicas, tras su admi-nistración intraduodenal (100).
Se han llevado a cabo estudios in vivo con nanopartículas lipídicas admi-
nistradas por vía oral conteniendo tobramicina (101, 102), clozapina (95), ri-fampicina, isoniazida y pirazinamida (103), camptotecina (104), en los que seha demostrado que los vehículos lipídicos son capaces de mejorar tanto labiodisponibilidad como las propiedades farmacocinéticas de éstas moléculas.
También en el caso de la administración oral de moléculas peptídicas se han
obtenido interesantes resultados. Así, nuestro grupo ha diseñado para tal fin na-nopartículas lipídicas recubiertas con polímeros hidrofílicos como el PEG o elquitosano (63, 105, 106) (Figura 4.2), demostrando que ambas eran captadas deun modo similar por parte de células Caco-2. Cuando estas nanopartículas con-teniendo calcitonina, se administraron por vía oral a ratas, las nanopartículas re-
cubiertas de quitosano lograron un marcado y prolongado efecto hipocalcémi-co, que no apareció sin embargo tras la administración de las recubiertas de PEG(105, 106) (Figura 4.3). La probable explicación de este efecto ha de relacio-narse tanto con la mucoadhesión del quitosano como con la apertura de las unio-nes intercelulares que el quitosano promueve.
Una de las vías exploradas para las nanopartículas lipídicas ha sido la pul-
monar. Concretamente, se ha estudiado su distribución tras ser aerosolizadas a
FIGURA 4.2. Fotografías obtenidas por microscopía electronica de transmision de nanopartículasde tripalmitina recubiertas de PEG (izquierda), nanopartículas de tripalmitina/Miglyol® 812recubiertas de PEG (medio) y nanopartículas de tripalmitina recubiertas de quitosano (derecha)
FIGURA 4.3. Niveles de calcemia en suero obtenidos tras la administración intragástrica de unasolución de calcitonina (CT Sol), de nanopartículas de tripalmitina conteniendo calcitoninarecubiertas de de quitosano (CS-LN) o de PEG (PEG-LN) (media ± e.e., n = 6); *diferenciassignificativas al comparar CS-LN con CT Sol o con PEG-LN, ·α< 0.05 (Tomado de ref. 105).
ratas mediante un nebulizador ultrasónico, o bien administradas por vía intra-traqueal (72, 107). Las nanopartículas, marcadas con 99mTc fueron seguidas me-diante gammagrafía, comprobándose su rápido acceso a nódulos linfáticos re-gionales, y sugiriéndose la fagocitosis por macrófagos alveolares como elmecanismo más probable de captura. La acumulación observada en nódulos lin-fáticos también ha hecho que las nanopartículas lipídicas sean propuestas comovehículos terapéuticos tras su liberación pulmonar. Administración cutánea y transdérmica
En los últimos años, se han comercializado algunos productos de aplicación
en cosmética, basados en las nanopartículas lipídicas (108). De hecho, de losdenominados portadores lipídicos nanoestructurados, considerados como la se-gunda generación de nanopartículas lipídicas, se encuentran en el mercado mun-dial más de 20 productos relacionados con la cosmética.
Considerando que para los liposomas, éste fue también el primer paso pre-
vio a su entrada en el mercado farmacéutico para otras aplicaciones terapéuti-cas, la situación puede ser prometedora para las nanopartículas lipídicas. Se hacomprobado además, mediante nanopartículas fluorescentes, que éstas puedenpenetrar en la piel, especialmente a través del espacio que rodea al folículo pi-loso, lo que ofrece grandes expectativas para el tratamiento del acné o patolo-gías como la alopecia (70).
Recientemente se ha estudiado la posibilidad de que las nanopartículas lipídi-
cas actuen como sistemas transdérmicos para moléculas como la melatonina (71). Los resultados demostraron la existencia de absorción significativa para dicha hor-mona cuando era aplicada tras su incorporación a nanopartículas lipídicas, que ade-más conseguían mantener niveles significativos durante un período de 24 horas.
Es una vía todavía poco explorada para la administración de nanopartícu-
las lipídicas, pero sin embargo el estudio realizado por el grupo de Gasco y col. (73) con tobramicina tópica crea nuevas expectativas en este área. Destaca es-pecialmente tanto el notable incremento observado en el AUC de las concen-traciones de fármaco en humor acuoso, que se multiplicó por un factor de 4, asícomo en el tmáx, que se multiplicó por 8, en comparación a la administracióndel colirio comercial. El seguimiento de estas nanopartículas marcadas con fluo-
rescencia explica los buenos resultados obtenidos, ya que tanto la retención anivel precorneal como a nivel del saco conjuntival se prolonga notablemente, loque es crucial en este tipo de administración. BIBLIOGRAFÍA
(1) Bangham, A.D., Standish, M.M. & Watkins, J.C. (1965) Diffusion of univalent
ions across lamellae of swollen phospholipids. J. Mol. Biol. 13: 238-252.
(2) Perrie, Y. (2008) Gregory Gregoriadis: Introducing liposomes to drug delivery. J.
(3) Fenske, D.B., Chonn, A. & Cullis, P.R. (2008) Liposomasl Nanomedicines : an
emerging field. Toxicologic Pathology. 36: 21-29.
(4) Ehrlich, P. (1906) Collected studies on Immunology. John Wiley, New York, pp
(5) Takeuchi, H., Matsui, Y., Sugihara, H., Yamamoto, H. & Kawashima, Y. (2005)
Effectiveness of submicron-sized chitosan-coated liposomes in oral administrationof peptide drugs. Int. J. Pharm. 303: 160-170.
(6) Wu, Z.H., Ping, Q.N., Wei, Y. & Lai, J.M. (2004) Hypoglycemic efficacy of chi-
tosan-coated insulin liposmes afeter oral administration in mice. Acta Pharmacol. Sin. 25: 966-972.
(7) Maurer, N., Fenske, D.B. & Cullis, P.R. (2001) Developments in liposomal drug
delivery systems. Expert Opin. Biol. Ther. 1: 923-947.
(8) Simard, P., Leroux, J.C., Allen, C. & Meyer, O. (2007) Liposomes for drug delivery.
En Nanoparticles for Pharmaceutical Applications. Domb AJ, Tabata Y, Ravi KumarMNV (eds). American Scientific Publishers, Stevenson Ranch California, pp 1-62.
(9) El Magharaby, G.M., Barry, B.W. & Williams, A.C. (2008) Liposomas and skin:
from drug delivery to model membranes. Eur. J. Pharm. Sci. 34: 203-222.
(10) Cevc, G. (1996) Transfersomes, liposomes and other lipid suspensions on the skin:
permeation enhancement, vesicle penetration and transdermal drug delivery. Crit. Rev. Ther. Drug Carrier Sys. 13: 257-388.
(11) Azarmi, S., Roa, W.H. & Lobenberg, R. (2008) Targeted delivery of nanoparticles
for the treatment of lung diseases. Adv. Drug Deliv. Rev. 60: 863-875.
(12) Lian, T. & Ho, R. (2001) Trends and developments in liposome drug delivery
systems. J. Pharm. Sci. 90: 667-680.
(13) Saunders, L., Perrin, J. & Gammack, D.B. (1962) Ultrasonic irradiation of some
phospholipid solutions. J. Pharm. Pharmacol. 14: 576-572.
(14) Kremer, J.M.H., Van de Esker, M.W.J., Pathmamanoharan, C. & Wiersema, P.H.
(1977) Vesicles of variable diameter prepared by a modified injection method. Bio-chemistry. 16: 3932-35.
(15) Szoka, F. & Papahadjopoulos, D. (1980) Comparative properties and methods
of preparation of lipid vesicles (liposomes). Ann. Rev. Biophys. Bioeng. 9: 467-508.
(16) Szoka, F., Olson, F., Heath, T., Vail, W., Mayhew, E. & Papahadjopoulos, D. (1980)
Preparation of unilamellar liposomes of intermediate size (0.1-0.2 microns) by acombination of reverse phase evaporation and extrusion through polycarbonatemembranes. Biochim. Biphys. Acta. 601: 559-571.
(17) Talsma, H., Ozer, A.Y., Van Bloois, L. & Crommelin, D.J. (1989) The size re-
duction of liposome with a high pressure homogenizer (Microfluidizer): charac-terization of prepared dispersions and comparison with conventional methods. Drug Dev. Ind. Pharm. 15: 197-207.
(18) Kirby, C. & Gregoriadis, G. (1984) Dehydration-rehydration vesicles: A simple
method for high yield drug entrapment in liposomes. Biotechnology. 2: 979-984.
(19) Li, C. & Deng, Y. (2004) A novel method for the preparation of liposomes: free-
ze drying of monophase solutions. J. Pharm. Sci. 93: 1403-1414.
(20) Storm, G. & Crommelin, D.J.A. (1998) Liposomes: quo vadis? Pharm. Sci. Tech-
(21) Klibanov, A.L., Maruyama, K., Beckerleg, A.M., Torchilin, V.P. & Huang, L.
(1990) Amphipatic polyethyleneglycols effectively prolong circulation time in li-posomes. FEBS Lett. 268: 235-237.
(22) Senior, J., Delgado, C., Fisher, D., Tilcock, C. & Gregoriadis, G. (1991) Influen-
ce of surface hydrophilicity of liposomes on their interaction with plasma proteinsand clearance from the circulation: studies with polyethyleneglycol-coated vesi-cles. Biochim. Biophys. Acta. 1062: 77-82.
(23) Immordino, M.L., Dosio, F. & Cattel, L. (2006) Stealth liposomes: review of the
basic science, rationale and clinical applications; existing and potential. Int. J. Na-nomed. 1: 297-315.
(24) Torchilin, V.P., Trubetskoy, V.S., Whitemen, K.R., Calceti, P., Feruti, P. & Vero-
nese, F.M. (1995) New synthetic amphiphilic polymers for the steric protection ofliposomes in vivo. J. Pharm. Sci. 84: 1049-1053.
(25) Lasic, D.D. & Templeton, N.S. (1996) Liposomes in gene therapy. Adv. Drug De-
(26) Torchilin, V.P. (2006) Recent approaches to intracellular delivery of drugs and
DNA and organelle targeting. Annu. Rev. Biomed. Eng. 8: 343-375.
(27) Schnyder, A., Krahenbuhl, S., Torok, M., Drewe, J. & Huwyler, J. (2004) Targe-
ting of skeletal muscle in vitro using biotinylated immunoliposomes. Biochim. J. 377: 61-67.
(28) Portney, N.G. & Ozkan, M. (2006) Nano-oncology: drug delivery, imaging and
sensing. Annal. Bioanal. Chem. 384: 620-630.
(29) Torchilin, V.P. (2005) Recent advances with lipomes as pharmaceutical carriers. Nat. Rev. Drug Discovery. 4: 145-160.
(30) Allen, T.M. & Cullis, P.R. 2004. Drug delivery systems: entering the mainstream.
(31) Andresen, T.L., Jensen, S.S. & Jorgensen, K. (2005) Advanced strategies in lipo-
somal cancer therapy: problems and prospect of active and tumor specific drugrelease. Prog. Lipid Res. 44: 68-97.
(32) Torrado, J.J., Espada, R., Ballesteros, M.P. & Torrado-Santiago, S. (2008) Am-
photericin B formulations and drug targeting. J. Pharm. Sci. 97: 2405-2425.
(33) Walsh, T.J., Yeldandi, M., McEvoy, M., Gonzalez, C., Chanock, S., Freijeld, A.,
Seibel, N.I., Whitcomb, P.O., Jarosinski, P., Boswell, G., Bekersky, I., Alak, A.,Buell, D., Barret, J. & Wilson, W. (1998) Safety, tolerance and pharmacokineticsof small unilamellar liposomal formulation of amphotericin B (Ambisome) in neu-tropenic patients. Antimicrob. Agents Chemother. 42: 2391-2398.
(34) Désormaux, A. & Bergeron, M.G. (2005) Lymphoid tissue targeting of anti-HIV
drugs using liposomes. Methods Enzymol. 391: 330-351.
(35) Gagné, J.F., Désormaux, A., Perron, S., Tremblay, M.J. & Bergeron, M.G. (2002)
Targeted delivery of indinavir to HIV-1 promary reservoirs with immunoliposo-mes. Biochim. Biophys. Acta. 1558: 198-210.
(36) Feldmann, M., Brennan, F.M. & Maini, R.N. (1996) Rheumathoid Arthritis. Cell.
(37) Tran, C.N., Lundy, S.K. & Fox, D.A. (2005) Synovial biology and T cells in rheu-
matoid arthritis. Pathophysiology. 12: 183-189.
(38) Vanniasinghe, A.S., Bender, V. & Manolios, N. The potential of liposomes drug
delivery for the treatment of inflammatory arthritis. Sem. Arthrit. Reuma. En pren-sa. doi: 10.1016/j.semarthrit.2008.08.004.
(39) Lee, L., Buckley, C., Blades, M.C., Panay, G., George, A.J. & Pitzalis, C. (2002)
Identification of synovium-specific homing peptides in vivo phage display sec-tion. Arthritis Rheum. 46: 2109-2120.
(40) Gaspar, M.M., Bakowsky, U. & Ehrhardt, C. (2008) Inhaled liposomes. Current
strategies and future challenges. J. Biomed. Nanotechnol. 4: 1-13.
(41) Zaru, M., Mourtas, S., Klepetsanis, A., Fadda, A.M. & Antimisiaris, S.G. (2007)
Liposomes for drug delivery to the lung by nebulization. Eur. J. Pharm. Biopharm. 67: 655-666.
(42) Bridges, P.A. & Taylor, K.M. (1998) Nebulisers for the generation of liposomal
aerosols. Int. J. Pharm.173: 117-125.
(43) Wong, J.P., Yang, H.M., Blasetti, K.L., Schnell, G., Conley, J. & Schofield, L.N.
(2003) Liposome delivery of ciprofloxacin against intracellular Francisella tula-rensis infection. J. Control. Release. 92: 265-273.
(44) Clark, J.M., Whitney, R.R., Olsen, S.J., George, R.J., Swerdel, L., Kunselman, L.
& Bonner, D.P. (1991) Amphotericin-B lipid complex therapy of experimental fun-gal- infections in mice. Antimicrobial Agents Chemother. 35: 615-621.
(45) Leemans, J.C., Juffermans, N.P., Florquin, S., Van Rooijen, N., Vervoordeldonk,
M.J., Verbon, A., Van Derenter, S.J.H. & Van der Poll, T. (2001) Depletion of Al-veolar Macrophages Exerts Protective Effects in Pulmonary Tuberculosis in Mice. J. Immunol. 166: 4604-4611.
(46) Chono, S., Tanino, T., Seki, T. & Morimoto, K. (2007) Uptake characteristics of
liposomes by rat alveolar macrophages: influence of particle size and surface man-nose modification. J. Pharm. Pharmacol. 59: 75-80.
(47) Khanna, C., Hasz, D.E., Klausner, J.S. & Anderson, P.M. (1996) Aerosol delivery
of interleukin-2 lipoosmes is non toxic and biologically effective: canine studies. Clin. Cancer Res. 2: 721-734.
(48) Khanna, C., Anderson, P.M., Hasz, D.E., Katsanis, E., Neville, M. & Klausner,
J.S. (1997) Interleukin2 liposome inhalation therapy is safe and effective for dogswith spontaneous pulmonary metastases. Cancer. 79: 1409-1421.
(49) Verschraegen, C.F., Gilbert, B.E., Loyer, E., Huaringa, A., Walsh, G., Newman,
R.A. & Knight, V. (2004) Clinical evaluation of the delivery and safety of aero-solized liposomal 9-nitro-20(S)-camptothecin in patients with advanced pulmo-nary malignancies. Clin. Cancer Res. 10: 2319-2326.
(50) Laube, B.L. (2005) The expanding role of aerosols in systemic drug delivery, gene
therapy and vaccination. Respir. Care. 50: 1161-1176.
(51) Huang, Y.Y. & Wang, C.H. (2006) Pulmonary delivery of insulin by liposomal ca-
rriers. J. Control Release. 113: 9-14.
(52) Kaur, I.P., Garg, A., Singla, A.K. & Aggarwal, D. (2004) Vesicular systems in ocu-
lar drug delivery: an overview. Int. J. Pharm. 269: 1-14
(53) Reddy, I.K. & Ganesan, M.G. (1996) Ocular therapeutics and drug delivery: a
multidisciplinary approach. Technomic Publishing Company.
(54) Sanchez, A. & Alonso, M.J. (2006) Nanoparticular carriers for ocular drug deli-
very. En: Nanoparticulates as drug carriers. Wei K (ed). World Scientific ImperialCollege Press.
(55) Al-Muhammed, J., Ozer, A.Y., Ercan, M.T. &, Hinkal, A.A. (1996) In vivo estudies
on dexamethasone sodium phosphate liposomes. J. Microencapsul. 13: 293-306.
(56) Ebrain, S., Peyman, G.A. & Lee, P.J. (2005) Applications of liposomes in oph-
thalmology. Surv. Ophthalmol. 50: 167-182.
(57) Dean, D.A., Byrd, J.N. & Dean, B.S. (1999) Nuclear targeting of plasmid DNA
in human corneals cells. Curr. Eye Res. 19: 66-75.
(58) Bochot, A., Couvreur, P. & Fattal, E. (2000) Intravitreal administration of anti-
sense oligonucleotides: potential of lipomal delivery. Prog. Retin. Eye Res. 19:131-147.
(59) Gasco, M.R. (1993) Method for producing solid lipid microspheres having a na-
rrow size distribution. U.S. Patent No 5.250.236.
(60) Müller, R.H. & Lucks, J.S. (1996) Arneistoffträger aus festen lipidteilchen, feste
lipidnanosphären (SLN)/ Medication vehicles made of solid lipid particles (solidlipid nanospheres - SLN). European Patent EP 0605497 B1.
(61) Gasco, M.R. (2007) Lipid nanoparticles: perspectives and challenges. Adv. Drug
(62) Manjunath, K., Reddy, J.S. & Venkateswarlu, V. (2005) Solid lipid nanoparticles
as drug delivery systems. Methods Find. Exp. Clin. Pharmacol. 27: 127-144.
(63) García-Fuentes, M., Torres, D. & Alonso, M.J. (2005) New surface-modified lipid
nanoparticles as delivery vehicles for salmon calcitonin. Int. J. Pharm. 296: 122-132.
(64) Wissing, S.A., Kaiser, O. & Müller, R.H. (2004) Solid lipid nanoparticles for pa-
renteral drug delivery. Adv. Drug Deliv. Rev. 56: 131-155.
(65) Shenoy, V.S., Vijay, I.K. & Murthy, R.S.R. (2005) Tumor targeting: biological fac-
tors and formulations advances in injectable lipid nanoparticles. J. Pharm. Phar-macol. 57: 127-144.
(66) Wong, H.L., Bendayanm R., Rauthm A.M., Lim Y. & Wu, X.Y. (2007) Chemo-
therapy with anticancer drugs encapsulated in solid lipid nanoparticles. Adv. DrugDeliv. Rev. 59: 491-504.
(67) Hu, L., Tang, X. & Cui, F. (2004) Solid Lipid Nanoparticles (SLNs) to improve
oral bioavailability of poorly soluble drugs. J. Pharm. Pharmacol. 56: 1527-1535.
(68) Muchow, M., Maincent, P. & Müller, R.H. (2008) Lipid nanoparticles with a so-
lid matrix (SLN®, NLC®, LDC®) for oral drug delivery. Drug Dev. Ind. Pharm. 34: 1394-1405.
(69) Müller, R.H., Radtke, M. & Wissing, S.A. (2002) Solid lipid nanoparticles (SLN)
and nanostructured lipid carriers (NLC) in cosmetic and dermatological prepara-tions. Adv. Drug Deliv. Rev. 54: 131-155.
(70) Wissing, S. & Müller, R.H. (2002) Solid lipid nanoparticles as carriers for suns-
creens: in vitro release and in vivo skin penetration. J. Control. Rel. 81: 225-233.
(71) Priano, L., Esposti, D., Esposti, R., Castagna, G., De Medici, C., Fraschini, F. &
Gasco, M.R. (2007) Solid lipid nanoparticles incorporating melatonin as new mo-del for sustained oral and transdermal delivery systems. J. Nanosci. Nanotechnol. 7: 1-6.
(72) Videira, M.A., Botelho, M.F., Santos, A.C., Gouveia, L.F., Pedroso de Lima, J.J.
& Almeida, A.J. (2002) Pulmonary delivery of SLN as a coloidal drug carriersystem to the lung lymphatics. J. Drug Target. 10: 607-613.
(73) Cavalli, R., Gasco, M.R., Chetoni, P., Burgalassi, S. & Saettone, M.F. (2002) So-
lid lipid nanoparticles (SLN) as ocular delivery system for tobramycin. Int. J. Pharm. 238: 241-245.
(74) Westensen, K., Bunjes, H. & Koch, M.H.J. (1997) Physicochemical characteriza-
tion of lipid nanoparticles and evaluation of their drug loading capacity and sus-tained release potential. J. Control Rel. 48: 223-236.
(75) Müller R.H., Mäder K., Lippacher A. & Jenning V. (2000) Fest-flüssige (halbfes-
te) Lipidpartikel und Verfahren zur Herstellung hochkonzentrierter Lipidpartikel-dispersionen. PCT application PCT/EP00/04565.
(76) Müller R.H., Mehnert W., Lucks J.S., Schwarz J.S., zur Mühlen, A., Weyhers, H.,
Freitas, C. & Rühl, D. (1995) Eur. J. Pharm. Biopharm. 41: 62-69.
(77) Sjöström, B. & Bergenstahl (1992) Preparation of submicron drug particles in le-
cithin-stabilized o/w emulsions, I. Model studies of the precipitation of choles-teryl acetate. Int. J. Pharm. 88: 53-62.
(78) Saraf, S., Mishra, D., Asthana, A., Jain, R., Singh, S. & Jain, N.K. (2006) Lipid
nanoparticles for mucosal immunization against hepatitis B. Vaccine. 24: 45-56.
(79) García-Fuentes, M., Torres, D. & Alonso, M.J. (2002) Design of lipid nanoparti-
cles for the oral delivery of hydrophilic macromolecules. Coll. Surf. B. Biointerf. 27: 159-168.
(80) Trotta, M., Cavalli, R., Carlotti, M.E., Battaglia, L. & Debernardi, F. (2005) So-
lid lipid micro-particles carrying insulin formed by solvent-in-water emulsion-dif-fusion technique. Int. J. Pharm. 288: 281-288.
(81) Hu, F.Q., Hong, Y. & Yuan, H. (2004) Preparation and characterization of solid
lipid nanoparticles containing peptide. Int. J. Pharm. 273: 29-35.
(82) Blasi, P., Giovagnoli, S., Schoubben, A., Ricci, M. & Rossi, C. (2007) Solid lipid
nanoparticles for targeted brain drug delivery. Adv. Drug Deliv. Rev. 59: 454-477.
(83) Yang, S.C., Lu, L.F., Cai, Y., Zhu, J.B., Liang, B.W. & Yang, C.Z. (1999) Body
distribution in mice of intravenously injected M.R. (2002). Intravenous adminis-tration to rabbits of non-stealth and stealth doxorubicin loaded lipid nanoparticlesat increasing concentrations of stealth agent: pharmacokinetics and distribution ofdoxorubicin in brain and in other tissues. J. Drug Target. 10: 327-335.
(84) Zara, G.P., Cavalli, R., Fundarò, A., Bargoni, A., Caputo, O. & Gasco, M.R. (1999)
Pharmacokinetics of doxorubicin incorporated in solid lipid nanospheres (SLN). Pharm. Res. 40: 281-286.
(85) Zara, G.P., Cavalli, R., Bargoni, A., Fundarò, A., Vighetto, D. & Gasco, M.R. In-
travenous administration to rabbits of non-stealth and stealth doxorubicin-loadedsolid lipid nanoparticles at increasing concentrations of stealth agent: pharmaco-kinetics and distribution of doxorubicin in brain and other tissues. J. Drug Target. 10: 327-335.
(86) Fundarò, A., Cavalli, R., Bargoni, A., Vighetto, D., Zara, G.P. & Gasco, M.R.
(2000) Non-stealth and stealth solid lipid nanoparticles (SLN) carrying doxorubi-cin: pharmacokinetics and tissue distribution after IV administration to rats. Pharm. Res. 42: 337-343.
(87) Reddy, L.H., Sharma, R.K., Chuttani, K., Mishra, A.K. & Murthy, R.S.R. (2005)
Influence of administration route on tumour uptake and biodistribution of etopo-side loaded solid lipid nanoparticles in Dalton´s lymphoma tumor bearing mice. J. Control. Rel. 105: 185-198.
(88) Serpe, L., Catalano, M.G., Cavalli, R., Ugazio, E., Bosco, O., Canaparo, R., Mun-
toni, E., Frairia, R., Gasco, M.R., Eandi, M. & Zara, G.P. (2004) Cytotoxicity ofanticancer drugs incorporated in solid lipid nanoparticles on HT-29 colorectal can-cer cell line. Eur. J. Pharm. Biopharm. 58: 673-680.
(89) Wong, H.L., Rauth, A.M., Bendayan, R., Manias, J.L., Ramaswamy, M., Liu, Z.,
Erhan, S.Z. & Wu, X.Y. (2006) A new polymer-lipid hybrid nanoparticle systemincreases cytotoxicity of doxorubicin against multidrug resistant human breast can-cer cells. Pharm. Res. 23: 1574-1585.
(90) Wong, H.L., Bendayan, R., Rauth, A.M., Xue, H.Y., Babakhanian, K. & Wu, X.Y.
(2006) A mechanistic study of enhanced doxorubicin uptake and retention in mul-tidrug resistant breast cancer cells using a polymer-lipid hybrid nanoparticle (PLN)system. J. Pharmacol. Exp. Ther. 317: 1372-1381.
(91) Wong, H.L., Rauth, A.M., Bendayan, R. & Wu, X.Y. (2007) In vivo evaluation of
a new polymer-lipid hybrid nanoparticle (PLN) formulation of doxorubicin in amurine solid tumor model. Eur. J. Pharm. Biopharm. 65: 300-308.
(92) Minagawa, T., Sakanaka, K., Inaba, S.I., Sai, Y., Tamai, I., Suwa, T. & Tsuji, A.
(1996) Blood-brain-barrier transport of lipid microspheres containing clinprost,a prostaglandin 12 analogue. J. Pharm. Pharmacol. 48: 1016-1022.
(93) Podio, V., Zara, G.P., Carazzone, M., Cavalli, R. & Gasco, M.R. (2000) Biodis-
tribution of stealth and non-stealth solid lipid nanospheres after intravenous ad-ministration to rats. J. Pharm. Pharmacol. 52: 1057-1063.
(94) Harivardhan, L., Reddy, L.H., Sharma, R.K., Chuttani, K., Mishra, A.K. &
Murthy, R.S.R. (2004) Etoposide-incorporated tripalmitin nanoparticles with dif-ferent surface charge: formulation, characterization, radiolabeling and biodistri-bution studies. AAPS J. 6 (Article 23, http:// www.aapsj.org)
(95) Manjunath, K. & Venkateswarlu, V. (2005) Pharmacokinetics, tissue distribution
and bioavailability of clozapine solid lipid nanoparticles after intravenous and in-traduodenal admininistration. J. Control Rel. 107: 215-228.
(96) Lockman, P.R., Koziara, J.M., Mumper, R.J. & Allen, D.D. (2004) Nanoparticle
surface changes alter blood-brain barrier integrity and permeability. J. Drug Tar-get. 12: 635-641.
(97) Charman, W.N. (2000) Lipids, lipophilic drugs, and oral drug delivery-some
emerging conceps. J. Pharm. Sci. 89: 967-978.
(98) Porter, C.J. & Charman, W.N. (2001) Intestinal lymphatic drug transport: An up-
date. Adv. Drug Deliv. Rev. 50: 61-80.
(99) Khoom S.M., Shacklefordm D.M., Porterm C.J., Edwardsm G.A. & Charmanm
W.N. (2003) Intestinal lymphatic trnaspot of halofantrine occurs after oral admi-nistration of a unit-dose lipid-based formulation to fasted dogs. Pharm. Res. 20:1460-1465.
(100) Bargoni, A., Cavalli, R., Caputo, O., Fundarò, A., Gasco, M.R. & Zara, G.P.
(1998) Solid lipid nanoparticles in lymph and plasma after duodenal administra-tion to rats. Pharm. Res. 15: 745-750.
(101) Cavalli, R., Zara, G.P., Caputo, O., Bargoni, A., Fundarò, A. & Gasco, M.R.
(2000) Transmucosal transport of tobramycin incorporated in solid lipid nano-particles (SLN) after duodenal administration. Part I- a pharmacokinetic study. Pharmacol. Res. 42: 541-545.
(102) Bargoni, A., Cavalli, R., Zara, G.P., Fundarò, A., Caputo, O. & Gasco, M.R.
(2001) Transmucosal transport of tobramycin incorporated in solid lipid nano-particles (SLN) after duodenal administration. Part II- tissue distribution. Phar-macol. Res. 43: 497-502.
(103) Pandey, R., Sharma, S. & Khuller, G.K. (2005) Oral solid lipid nanoparticle- ba-
sed antitubercular chemotherapy. Tuberculosis. 85: 415-420.
(104) Yang, S.C., Zhu, J.B., Lu, Y., Liang, B.W. & Yang, C.Z. (1999) Body distribu-
tion of camptothecin solid lipid nanoparticles after oral administration. Pharm. Res. 16: 751-757.
(105) García-Fuentes, M. (2004) Nanopartículas lipídicas modificadas con polímeros
hidrófilos: desarrollo y evaluación de su potencial como sistemas transportado-res de péptidos a nivel intestinal. Universidad de Santiago de Compostela.
(106) García-Fuentes, M., Prego, C., Torres, D. & Alonso, M.J. (2005) A comparative
study of the potential of solid triglyceride nanostructures coated with chitosan orpoly(ethyleneglycol) as carriers for oral calcitonin delivery. Eur. J. Pharm. Sci. 25: 133-143.
(107) Videira, M., Gano, L., Santos, A.C., Neves, M. & Almeida, A.J. (2006) Lympha-
tic uptake of lipid nanoparticles following endotracheal administration to rats. J. Microencapsul. 23: 855-862.
(108) Müller, R.H., Rimpler, C., Petersen, R., Hommoss, A. & Schwabe, K. (2007) A
new dimension in cosmetic products by nanostructured lipid carriers (NLC) tech-nology. Eur. Cosmet. 15: 32-37.
Veterinary PrODUCtS liSt Contacts : Tel +33 (0) 1 55 46 50 00 - Fax +33 (0) 1 47 39 91 90jpinguet@quimdis.com - odelasayette@quimdis.com - igohourou@quimdis.com - michel.manalt@quimdis.comThe availability of these products are under the local legislation. Patent position to be verified by the customer. 4-AMINO BENZOIC ACID CLOSANTEL SODIUM ACETIC ACID COBALT GLUCONATE ACETYLCY
NADINE JURDI ALAEDDINE – Page 1 of 5 N A D I N E J U R D I A L A E D D I N E PERSONAL PROFILE I am a well respected, dedicated and hard working professional with diverse experience across many areas. I am recognized for my ability to achieve results due to exceptional interpersonal, analytical and problem solving skills, as well as the ability to be detail oriented and multi task eff
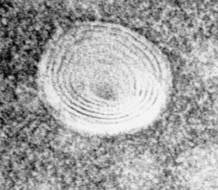 4. Nanosistemas lipídicos
4. Nanosistemas lipídicos
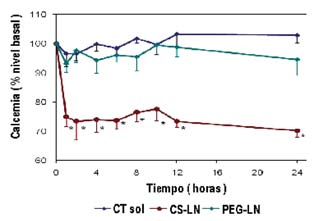 cubiertas de quitosano lograron un marcado y prolongado efecto hipocalcémi-co, que no apareció sin embargo tras la administración de las recubiertas de PEG(105, 106) (Figura 4.3). La probable explicación de este efecto ha de relacio-narse tanto con la mucoadhesión del quitosano como con la apertura de las unio-nes intercelulares que el quitosano promueve.
cubiertas de quitosano lograron un marcado y prolongado efecto hipocalcémi-co, que no apareció sin embargo tras la administración de las recubiertas de PEG(105, 106) (Figura 4.3). La probable explicación de este efecto ha de relacio-narse tanto con la mucoadhesión del quitosano como con la apertura de las unio-nes intercelulares que el quitosano promueve.