La tétracycline, connue sous le nom commercial Sumycin, agit en bloquant la fixation de l’ARNt sur la sous-unité 30S ribosomale, interrompant l’élongation de la chaîne protéique bactérienne. Ce mécanisme confère une activité sur un spectre large, incluant bactéries Gram positives, Gram négatives, rickettsies et spirochètes. Sa biodisponibilité digestive varie selon la prise alimentaire et les interactions avec les ions divalents comme calcium et magnésium. Sa diffusion tissulaire est importante, notamment dans les voies respiratoires et génito-urinaires. L’élimination se fait par voie rénale et biliaire. Les effets indésirables incluent photosensibilisation, troubles digestifs et coloration dentaire en cas d’administration précoce. Les guides thérapeutiques mentionnent sumycin prix, en soulignant la nécessité de restreindre son utilisation afin de limiter les résistances acquises.
Static0.volkskrant.nl
J Clin Endocrin Metab. First published ahead of print April 30, 2013 as doi:10.1210/jc.2012-3893 Molecular Diagnosis of 5␣-Reductase Deficiency in 4 Elite Young Female Athletes Through Hormonal Screening for Hyperandrogenism
Patrick Fénichel, Françoise Paris, Pascal Philibert, Sylvie Hiéronimus, Laura Gaspari,Jean-Yves Kurzenne, Patrick Chevallier, Stéphane Bermon, Nicolas Chevalier, andCharles Sultan
Department of Reproductive Endocrinology and Institut National de la Santé et de la Recherche MédicaleUnité 1065 (P.F., S.H., N.C.), Hospital of L’Archet, University Hospital of Nice, 06200 Nice, France;Department of Hormonology and Pediatric Endocrinology (F.P., P.P., L.G., C.S.), Lapeyronie Hospital,Centre Hospitalier Universitaire, 34295 Montpellier, France; Departments of Pediatrics (J.-Y.K.) andMedical Imaging (P.C.), Centre Hospitalier Universitaire Nice, France; and Monaco Institute for SportMedicine and Surgery (IM2S) (S.B.), 98000 Monaco
Context: Although a rare occurrence, previously undiagnosed disorders of sex development (DSD) with hyperandrogenism are sometimes detected by hormonal screening during the international sports competitions. Identifying the cause of XY,DSD raises medical and ethical concerns, especially with regard to issues of the eligibility to compete. Objective: The aim of this study was to determine whether the detection of high plasma T in young elite female athletes during hormonal screening would reveal an unsuspected XY DSD. Setting: The study was performed in the Nice and Montpellier University Hospitals (France), which collaborate as reference centers for DSD in elite athletes on behalf of sports governing bodies. Patients: Four cases of elite young athletes with female phenotypes but high plasma T detected during hormonal screening were investigated for undiagnosed XY DSD. Main Outcome Measures: Evaluation of clinical, biological, radiological (magnetic resonance im- aging and dual-energy x-ray absorptiometry) and genetic characteristics was conducted. Results: The 4 athletes presented as tall, slim, muscular women with a male bone morphotype, no breast development, clitoromegaly, partial or complete labial fusion, and inguinal/intralabial tes- tes. All reported primary amenorrhea. The hormonal analysis evidenced plasma T within the male range, the karyotype was 46, XY, and molecular analysis of the 5␣-reductase type 2 (srd5A2) gene identified a homozygotic mutation in 2 cases, a heterozygotic compound in 1 case, and a deletion in 1 case. Conclusion: 5␣-Reductase deficiency should be investigated in elite young female athletes with primary amenorrhea and high male T levels detected during antidoping programs to identify undiagnosed XY DSD. Theworldofsportshasstruggledwiththeissueofgen- versial.Themajorquestioniswhetherthisconditionpro-
der abnormalities since the Olympic Games of Berlin
vides unfair advantages. The focus is not so much on
in 1936 (1). The matter of systematic screening for ab-
uncovering cases of masquerading or anabolic doping but
normal virilization in female athletes remains still contro-
on detecting those athletes who are competing unknow-
Abbreviations: CAIS, complete androgen insensitivity syndrome; DHT, dihydrotestoster-
one; DSD, disorder of sex differentiation; MRI, magnetic resonance imaging; SRD5A2,
Copyright 2013 by The Endocrine Society
deficiency in 5␣-reductase type 2.
Received November 13, 2012. Accepted April 16, 2013. Copyright (C) 2013 by The Endocrine Society
5␣-Reductase Deficiency and Female Athletes
ingly with a disorder of sex differentiation (DSD). A sys-
bone mineral density and corporeal composition (com-
tematic gender verification program was first established
parison with a female control population).
in 1966, with clinical inspection, Barr body screening, and
Molecular analysis of the SRD5A2 gene was performed
later sex-determining region, Y chromosome DNA detec-
in the Department of Hormonology of the University Hos-
tion by PCR, a decision that created controversy and
pital of Montpellier. Genomic DNA was extracted from
caused considerable embarrassment (2). The decision to
peripheral blood leukocytes following the manufacturer’s
abandon compulsory gender verification in Olympic com-
instructions (DNA QIAamp DNA blood minikit; QIA-
petition was made in 1999. Today the new systematic
GEN, Courtaboeuf, France). As previously reported (6),
hormonal screening in phenotypically female athletes (3)
exons 1–5 of the SRD5A2 gene were amplified by PCR,
may be a good opportunity to identify unknown XY DSD
and direct sequencing was performed using the BigDye
with primary amenorrhea and hyperandrogenism.
terminator version 1.1 kit (Applied Biosystems, Courta-
Partial androgen insensitivity syndrome or minimal an-
boeuf, France) and an AB1 Prism 310 genetic analyzer
drogen insensitivity syndrome may be implicated, but not
(Applied Biosystems). For this retrospective clinical study,
complete androgen insensitivity syndrome (CAIS), be-
institutional review board/International Electrotechnical
cause in patients with CAIS, plasma T is very high but
Commission approval for publication is not required in
inactive. Another cause of XY DSD with possible hyperan-
drogenic amenorrhea is a deficiency in 5␣-reductase type2 (SRD5A2) activity. This is a very rare disorder charac-terized by high phenotypical variability. The spectrum ac-
tually ranges from isolated micropenis or hypospadias tosevere undervirilization appearing as normal female ex-
The 4 young amenorrheic women, respectively, 18, 20, 21,
ternal genitalia with mild clitoral enlargement (4). The
and 20 years of age at diagnosis, were from rural or moun-
diagnosis is made either at birth or at puberty when there
tainous regions of developing countries. They had never
is virilization on the external genitalia through either the
menstruated and this primary amenorrhea had never been
androgen receptor binding of very high levels of serum T,
evaluated. Consanguinity was confirmed for 3 of them
albeit at lower affinity, or the increased expression of ex-
(first cousins in cases 2 and 3 and siblings in case 4) and
tragenital 5␣-reductase type 1, which results in low pe-
was suspected in the fourth case (case 1), with the 2 parents
ripheral synthesis of dihydrotestosterone (DHT) from T
originating from neighboring villages (Table 1). All had
several brothers and sisters, including 1 sister who had
We report for the first time the diagnosis of 5␣-reduc-
been surgically treated for a DSD (nonverified; case 2) and
tase deficiency in 4 hyperandrogenic elite female athletes.
1 infertile (case 3) (Table 1). They all reported unexpectedvirilization at puberty with excessive pubic hair or clitoro-megaly. In all cases, they had manifested strong motiva-
Patients and Methods
tion and high tolerance to intensive daily training, whichhad made them good candidates for elite sports competi-
Four elite young amenorrheic athletes with hyperandro-
tion. However, none of them reported male sex behavior.
genism were referred to our Reproductive Endocrinology
The clinical characteristics were quite similar, with all
Department. Case 1 was identified through an abnormal
presenting as tall, slim, and muscular young women but
urine steroid profile and clitoral hypertrophy reported by
with a complete lack of breast development and android
the antidoping officer, whereas cases 2 and 4 through in-
bone morphotype (high biacromial/bitrochanteric diam-
creased plasma T, and free androgen index and LH results
eter ratio) (Table 1). Axillary hair growth was slight and
collected as a part of the Athlete Biological Passport (3).
pubic hair was female triangular, and none presented with
Case 3 was directly referred to the international federation
hirsutism. Clinical inspection of the external genitalia re-
medical department by her national federation doctor.
vealed clitoromegaly in all cases, almost complete labial
Blood samples were taken for endocrine investigation
fusion in cases 2 and 4, partial fusion in cases 1 and 3, and
and for karyotyping and genetic analysis after informed
a single urogenital orifice in cases 2 and 4 (types 2– 4 of the
consent was obtained. Steroids and polypeptide hormones
Prader classification) (Table 1). Clinical palpation of the
were assayed by radioimunological methods currently
major labia or inguinal region and MRI of the abdominal-
used in the University Hospital of Nice. Abdominal-pelvic
pelvic region determined the nature and size of the testes
magnetic resonance imaging (MRI) was systematically
and localized them at the inguinal orifice in cases 1 and 2,
performed, as was dual-energy x-ray biphotonic absorp-
intralabial in case 4, and 1 at the inguinal orifice and the
tiometry (Hologic, Bedford, Massachusetts) to determine
other sublabial in case 2 (Table 1). MRI confirmed the lack
Height/Weight, Age at Diagnosis, y Consanguinity Bioacromial/Bitrochanteric Prader Class
Total T, ng/mL [0.09 – 0.99] DHT, ng/mL T to DHT Ratio D4, ng/mL [0.4 –2.3] 17 OHP, ng/mL [0.1–2.9] E2, pg/mL [19 –214] LH,
Abbreviations: AMH, anti-Mullerian hormone; DEXA, dual-energy x-ray absorptiometry; E2, estradiol; 17OHP, 17-hydroxyprogesterone.
a Female ratio 1, 1; male ratio 1, 3 (18).
of Mullerian ducts and the presence of intrapelvic seminal
and high T levels prompted the molecular analysis of the
vesicles and a rudimentary prostate in all athletes, whereas
SRD5A2 gene by direct sequencing, which revealed a ho-
a short blinded vagina (Ͻ22 mm) was identified in cases 1,
mozygous mutation in 2 cases in exon 1 (p.Gly34Arg; case
2, and 3. In addition, the vertebral bone mineral content
1) and in exon 4 (p.Asn.193Ser; case 2), a compound
was decreased in cases 1 and 2 compared with standards
(p.[Arg227X]ϩ[Ala228Thr]), and an exon 1 deletion in
Table 1 summarizes the hormonal features, showing a
case 3 (Figure 1). Regarding the compound heterozygous
male range of basal plasma total T with elevated gonad-
mutation (case 4), we confirmed by direct sequencing that
otropins and normal male estradiol, 17-hydroxyproges-
1 mutation was inherited from the father, whereas the
terone, anti-Mullerian hormone, and inhibin B. DHT was
other was inherited from the mother.
decreased with an increased T to DHT ratio except for case
In contrast to the tendency to request gender change,
our 4 athletes wished to maintain their female identity and
Chromosomal analysis confirmed a 46, XY karyotype.
had many questions about menstruation, sexual activity,
The combination of primary amenorrhea, lack of breast
and child-bearing. Although leaving male gonads in
development, clitoromegaly, a female pubic hair pattern,
SDRD5A2 patients carries no health risk, each athlete was
Figure 1. Sequencing electrophoregrams showing the different mutants identified in the 4 athletes with SRD5A2 deficiency.
5␣-Reductase Deficiency and Female Athletes
informed that gonadectomy would most likely decrease
However, it is possible that the gender abnormalities of
their performance level but allow them to continue elite
these athletes were clearly recognized at birth, especially
sport in the female category. We thus proposed a partial
for cases 2 and 4, but not formally diagnosed or given
clitoridectomy with a bilateral gonadectomy, followed by
medical attention because they had been born in rural
a deferred feminizing vaginoplasty and estrogen replace-
regions of countries with poor care.
ment therapy, to which the 4 athletes agreed after in-
The estimated incidence of severe 46, XY DSD with
formed consent on surgical and medical procedures.
uncertain sex is 2.2 per 10 000 births (15), and SRD5A2
Sports authorities then allowed them to continue compet-
deficiency accounts for 6.7% of such cases (16), for a set
ing in the female category 1 year after gonadectomy.
incidence of 1 per 100 000 (0.001%). In 1996 the system-atic sex-determining region, Y chromosome screening infemale athletes (2) during the Atlanta Olympic Games re-
Discussion
vealed 8 46XY DSD cases of 3384 (1 of 423) including 1case (1 of 3384) of SRD5A2 deficiency (known to but
We report here for the first time the cases of 4 elite young
hidden by the athlete herself). More recently, during a
amenorrheic athletes who were diagnosed with SRD5A2
World Championship, a massive systematic blood testing
deficiency after high androgen levels and have been re-
program was for the first time conducted: 868 top female
vealed during implementation of an antidoping program.
level athletes participated in this implementation of the
In these 4 46, XY DSD cases with female phenotype and
Athlete Biological Passport (3). Two cases of hyperandro-
high plasma T, the lack of breast development suggested
genism led to the diagnosis of SRD5A2 deficiency for an
SRD5A2 deficiency, even though DHT was not com-
incidence of at least 1 of 434 (some cases are still pending),
pletely abolished, as reported in our previous experience
which is more than 200 times the estimated incidence in
(7) or by others (8). We identified 2 known homozygous
the general population. These unpublished preliminary
missense mutations of SRD5A2 (p.Asn193Ser and
results suggest that the high active T during the fetal, post-
p.Gly34Arg), 1 new complete exon 1 deletion, and a com-
natal, or peripubertal period in cases of SRD5A2 defi-
ciency may confer a selective advantage for sports, likely
through the brain, muscle, and/or bone morphotype.
To our knowledge, the complete exon 1 deletion, iden-
The bone morphotype in our patients was android but,
tified in case 3, has never been reported. Conversely, the
curiously, the vertebral bone mineral content was de-
substitution of asparagine by serine at position 193
creased in 2 patients compared with standards for females.
(p.Asn193Ser) identified in homozygous form in case 2
One of the main issues that has emerged with regard to
was reported by our group in a 16-year-old Polish girl who
46, XY DSD hyperandrogenic athletes is the issue of pro-
exhibited clitoral enlargement and virilization (6). Simi-
tecting their health, private life, integrity, and rights but
larly, the p.Gly34Arg mutation found in case 1 was de-
maintaining strict standards of fairness for all women.
scribed in a 35-year-old Vietnamese woman with primary
This has led to the guidelines proposed by the Interna-
amenorrhea, no breast development, and clitoromegaly
tional Association of Athletics Federations
(8, 9). Regarding the compound heterozygous mutation
p.[Arg227X]ϩ[Ala228Thr] (case 4), the former mutation
national Olympic Committee and recently commented in
was reported in homozygous form in a 16-year-old Mex-
this journal (17). Except for cases of CAIS with inactive T,women with 46XY DSD will be allowed to compete only
ican girl with no breast development, primary amenorrhea
when T blood levels decrease on less than 10 nmol/L, the
and virilization (8) and in a 6-year-old boy with micrope-
nis, posterior hypospadias, but no cryptorchidism (9),
In this work, we demonstrate that young competitive
whereas the latter mutation was reported in homozygous
female athletes may be affected by an SRD5A2 deficiency.
form in a patient of Eritrean origin who presented at birth
It is thus important to screen for SRD5A2 deficiency in all
with perineal hypospadias, a hypoplastic scrotum with
young athletes with primary amenorrhea and hyperan-
both gonads palpable in the inguinal canal, and a micro-
drogenism to protect their health and privacy and ensure
The delay in diagnosing these athletes may be explained
by the slight degree of genital malformation generally ob-served in such cases at birth. Virilization then occurs at
Acknowledgments
puberty due to the rise in T and the 5␣-reductase type Ienzyme (11–13), with a possible female-to-male gender
Address all correspondence and requests for reprints to: Patrick
switch and/or a marked behavioral change (7, 13, 14).
Fenichel, MD, PhD, Researcher, Institut National de la Santé et
de la Recherche Médicale and Nice University Department of
10. Hiort O, Sinnecker GH, Willenbring H, Lehners A, Zöllner A, Struve D. Nonisotopic single strand conformation analysis of the
5␣-reductase type 2 gene for the diagnosis of 5␣-reductase defi-
Disclosure Summary: The authors have nothing to declare.
ciency. J Clin Endocrinol Metab. 1996;81:3415–3418.
11. Imperato-McGinley J, Peterson RE, Gautier T, Sturla E. Androgens
and the evolution of male-gender identity among male pseoudoher-maphrodistes with 5␣-reductase déficience. N Engl J Med. 1979;
References
12. Thiele S, Hoppe U, Holterhus PM, Hiort O. Isoenzyme type 1 of
1. Ritchie RJ, Reynard J, Lewis T. Intersex and the Olympic Games. R
5␣-reductase is abundantly transcribed in normal human genital
skin fibroblasts and may play an important role in masculinization
2. Elsas LJ, Ljungqvist A, Ferguson-Smith MA, et al. Gender verifica-
of 5␣-reductase type 2 deficient males. Eur J Endocrinol. 2005;152:
tion of female athletes. Genet Med. 2000;2:249 –254.
3. Sottas PE, Vernec A. Current implementation and future of the Ath-
13. Houk CP, Dzamiani D, Lee PA. Choice of gender in 5␣ reductase-2
lete Biological Passport. Bioanalysis. 2012;13:1645–1652.
deficiency: a moving target. J Pediatr Endocrinol Metab. 2005;18:
4. Maimoun L, Philibert P, Cammas B, et al. Phenotypical, biological,
and molecular heterogeneity of 5␣-reductase deficiency: an exten-
14. Mendonca BB, Inacio M, Costa EM, Arnhold IJ, Silva FA, Nicolau
sive international experience of 55 patients. J Clin EndocrinolW. Male pseudohermaphrodism due to steroid 5␣ reductase 2 de-
ficiency. Diagnosis, psychological evaluation and management.
5. Azzouni F, Godoy A, Li Y, Mohler J. The 5 alpha-reductase isozyme Medicine (Baltimore). 1996;75:64 –76.
family: a review of basic biology and their role in human diseases
15. Thuyen U, Lanz K, Holterhus PM, Hiort O. Epidemiology and ini-
tial management of ambiguous genitalia at birth in Germany. Horm
6. Boudon C, Lumbroso S, Lobacaro JM, et al. Molecular study of the
5␣-reductase type 2 gene in three European families with 5␣-reduc-
tase déficience. J Clin Endocrinol Metab. 1995;80:2149 –2153.
16. Erdog˘an S, Kara C, Uçaktürk A, Aydın M. Etiological classification
7. Maimoun L, Philibert P, Bouchard P, et al. Primary amenorrhea in
and clinical assessment of children and adolescents with disorders of
four adolescents revealed 5␣-reductase deficiency confirmed by mo-
sex development. J Clin Res Pediatr Endocrinol. 2011;3:77– 83.
lecular analysis. Fertil Steril. 2011;95:804.e1–5.
17. Xavier NA, McGill JB. Hyperandrogenism and intersex controver-
8. Johnson L, George FW, Neaves WB, et al. Characterization of the
sies in women’s Olympics. J Clin Endocrinol Metab. 2012;97:3902–
testicular abnormality in 5␣-reductase deficiency. J Clin Endocrinol
18. Vague J. Explorations clinique des syndromes testiculaires in “No-
9. Thigpen AE, Davis DL, Milatovich A, et al. Molecular genetics of tions d’Endocrinologie.” Paris: Jean Vague (Flammarion); 1965:
steroid 5␣-reductase 2 deficiency. J Clin Invest. 1992;90:799 – 809.
Section of Paediatric Oncology ICR Section of Paediatric Oncology, HaddowLaboratories, Sutton•Phase I/II clinical trial assessment of newPaediatric Oncology Unit, Sutton, including Relevance to the NHS Research and Development Programme strategies is a stated NHS priority area. As aleading group in the UK in investigational Chairman •development of techniques for molecularSecti
 J Clin Endocrin Metab. First published ahead of print April 30, 2013 as doi:10.1210/jc.2012-3893
J Clin Endocrin Metab. First published ahead of print April 30, 2013 as doi:10.1210/jc.2012-3893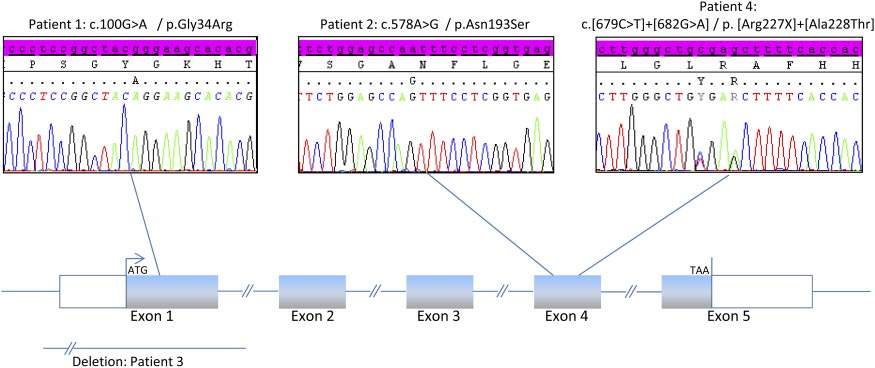 Height/Weight,
Height/Weight,