La tétracycline, connue sous le nom commercial Sumycin, agit en bloquant la fixation de l’ARNt sur la sous-unité 30S ribosomale, interrompant l’élongation de la chaîne protéique bactérienne. Ce mécanisme confère une activité sur un spectre large, incluant bactéries Gram positives, Gram négatives, rickettsies et spirochètes. Sa biodisponibilité digestive varie selon la prise alimentaire et les interactions avec les ions divalents comme calcium et magnésium. Sa diffusion tissulaire est importante, notamment dans les voies respiratoires et génito-urinaires. L’élimination se fait par voie rénale et biliaire. Les effets indésirables incluent photosensibilisation, troubles digestifs et coloration dentaire en cas d’administration précoce. Les guides thérapeutiques mentionnent sumycin prix, en soulignant la nécessité de restreindre son utilisation afin de limiter les résistances acquises.
No job name
J. Org. Chem. 1999, 64, 3778-3782 First Synthesis of Marine Sponge Alkaloid Niphatoxin B
Alexander Kaiser,*,† Christian Marazano, and
¨ r Pharmazie, Pharmazeutische Chemie I,Universita¨t Regensburg, D-93040 Regensburg, Germany
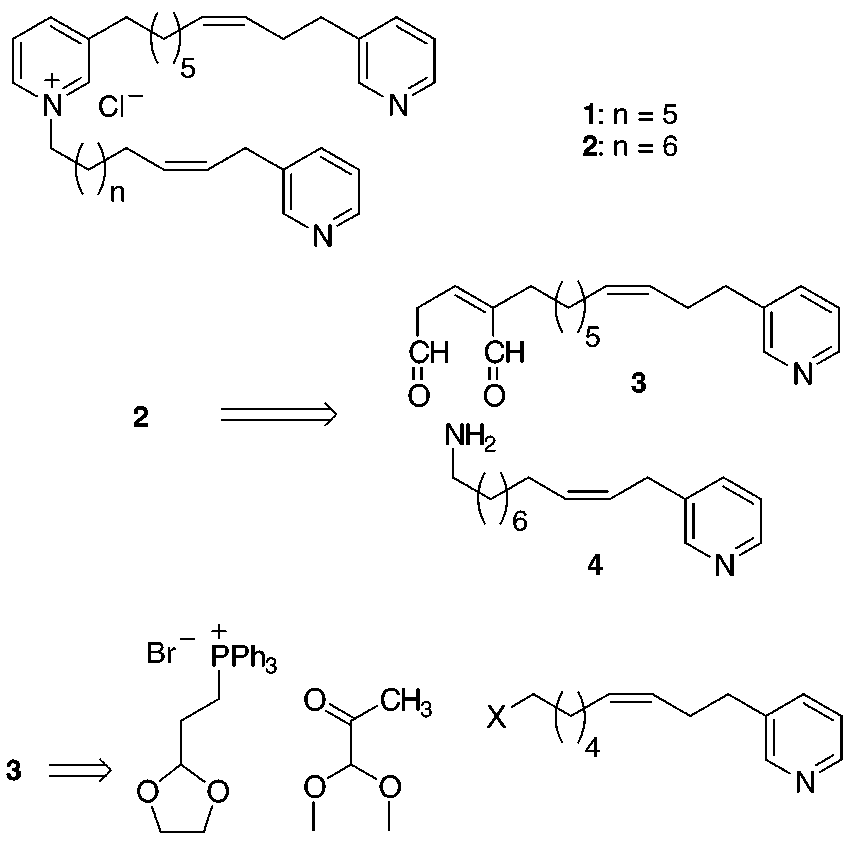
In 1992, Talpir et al. reported the isolation and
structure elucidation of niphatoxins A (1) and B (2), two ichthyo- and cytotoxic (IC
dine alkaloids.1,2 These Red Sea sponge alkaloids, isolatedfrom a Niphates sp., belong to an emerging and intrigu-ing class of marine secondary metabolites which arerelated to each other by their apparent biogenetic originfrom 3-alkylpyridine or reduced 3-alkylpyridine units.3Among them niphatoxins are unique in having threepyridine units and a substructure in which two pyridinerings are linked through their 3 positions to the samealkyl chain. Whereas other members of this group havebecome subjects of total syntheses or synthetic studies,4
commercially available three carbon fragments and a
no synthesis of niphatoxins has been reported up to now.
3-alkylpyridine unit. Na+- and K+-salts of glutaconalde-
Herein we report the first total synthesis of niphatoxin
hyde enolates have been used as starting materials to
B (2) based on a novel approach to glutaconaldehyde
prepare pyridines by reaction with ammonium salts.5
bisacetals.5 The retrosynthetic analysis for our approach
These glutaconaldehyde salts, however, have been ob-
is outlined in Scheme 1. Disconnections of the two C-N
tained from pyridine by ring opening reactions, and not
bonds in the pyridinium moiety lead to a 3-(ω-aminoalkyl)-
from acyclic starting materials. In a new biogenetic
pyridine 4 and 2-substituted glutaconaldehyde 3 which
hypothesis, amino derivatives of glutaconaldehyde were
in turn could be divided via its bisacetal into two
proposed as key intermediates in the biosynthesis ofmanzamine alkaloids.6
Our efforts were first directed to exploring the feasibil-
E-mail: alexander.kaiser@chemie.uni-regensburg.de.
(1) Talpir, R.; Rudi, A.; Ilan, M.; Kashman, Y. Tetrahedron Lett.
ity of the envisioned key reactions with simple model
1992, 33, 3033-3034.
compounds. Phosphonium salt 5 was deprotonated with
(2) The structural formula given in ref 1 on page 3034 appears to
n-BuLi in THF at -20 °C, and the resulting ylide was
be incorrect in respect to the molecular formulas in the text. Structuralformula given on page 3034 corresponds to molecular masses of 524
reacted with 1,1-dimethoxy-2-propanone (6) to give ole-
and 538 u, whereas molecular masses given in the text are 510 and
fination product 7 in 54% yield as a 9:1 mixture of E/Z-
524 u. Comparison of the masses of pyridinealkyl fragments resulting
isomers. Bearing in mind the low stability of glutacon-
from cleavage of the C-N bond in the text (216 and 230) with themasses of these units in the structural formula (230 and 244) shows
aldehyde in its free form,5 we planned to subject
that one methylene unit in the N+-alkyl chain should be omitted. The
intermediate 3 to the cyclocondensation with the amine
same incorrect structural formula appears also in ref 3. 4 without prior isolation. Acid treatment of 7 until
(3) For an excellent recent review, see: Andersen, R. J.; Van Soest,
R. W. M.; Kong, F. Alkaloids: Chemical and Biological Perspectives;
disappearance of the starting material (TLC), addition
Pelletier, S. W., Ed.; Pergamon Press: Elsevier Science: Oxford, U.K.,
of cyclohexylamine and Et3N, and reflux in n-butanol
(4) Manzamines, ircinol A, and ircinal A: (a) Winkler, J. D.; Axten,
gave disappointing results, probably due to decomposition
J. M. J. Am. Chem. Soc. 1998, 120, 6425-6426. (b) Magnier, E.;
of the resulting 2-methylglutaconaldehyde under the
Langlois, Y. Tetrahedron 1998, 54, 6201-6258 and references therein.
conditions of acetal hydrolysis. Finally, short treatment
Xestospongines: (c) Baldwin, J. E.; Melman, A.; Lee, V.; Firkin, C. R.; Whitehead, R. C. J. Am. Chem. Soc. 1998, 120, 8559-8560 and
of 7 with FeCl3,7 adsorbed on silica gel, in CH2Cl2 prior
references therein. Petrosines: (d) Heathcock, C. H.; Brown, R. C. D.;
to addition of cyclohexylamine hydrochloride and Et3N,
Norman, T. C. J. Org. Chem. 1998, 63, 5013-5030 and references
removal of CH2Cl2, and reflux in n-butanol afforded
therein. Sarains: (e) Downham, R.; Ng, F. W.; Overman, L. E. J. Org. Chem. 1998, 63, 8096-8097 and references therein. Mandangamin:
pyridinium salt 8 in 39% yield after column chromatog-
(f) Matzanke, N.; Gregg, R. J.; Weinreb, S. M. J. Org. Chem. 1997, 62,
1920-1921. Cyclostellettamines: (g) Kaiser, A.; Billot, X.; Gateau-
We next turned our attention to the preparation of the
Olesker, A.; Marazano, C.; Das, B. C. J. Am. Chem. Soc. 1998, 120, 8026-8034. (h) Baldwin, J. E.; Spring, D. R.; Atkinson, C. E.; Lee, V.
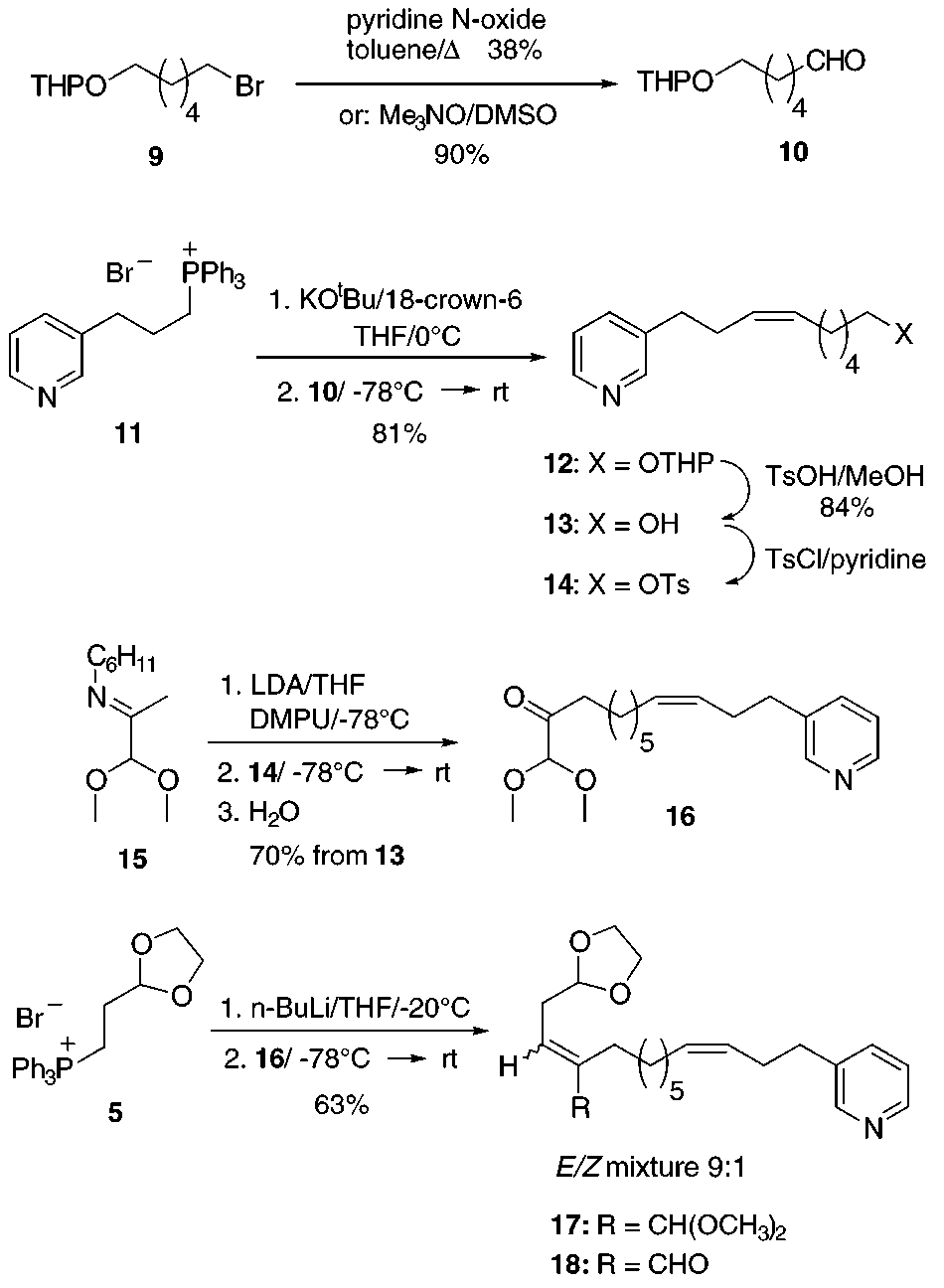
required glutaconaldehyde bisacetal. Known aldehyde8
Tetrahedron 1998, 54, 13655-13680 and references therein. Hal- 10 was prepared in 38% and 90% yields by oxidation of
iclamines: (i) Morimoto, Y.; Yokoe, C.; Kurihara, H.; Kinoshita, T.
THP-protected bromo alcohol 9 using pyridine N-oxide Tetrahedron 1998, 54, 12197-12214 and references therein. Halicy- clamine A and keramaphidine B: (j) Baldwin, J. E.; Claridge, T. D.
or trimethyamine N-oxide,9 respectively. (Z)-Selective
W.; Culshaw, A. J.; Heupel, F. A.; Lee, V.; Spring, D. R.; Whitehead,
Wittig olefination of aldehyde 10 with phosphonium salt
R. C.; Boughtflower, R. J.; Mutton, I. M.; Upton, R. J. Angew. Chem. 1998, 110, 2806-2808 and references therein. Niphatesines and theonelladins: (k) Bracher, F.; Papke, T. Monatsh. Chem. 1996, 127,
91-95 and references therein. (l) Teubner, A.; Gerlach, H. Liebigs Ann.
(7) Kim, K. S.; Song, Y. H.; Lee, B. H.; Hahn, C. S. J. Org. Chem.Chem. 1993, 161-165. Polymeric 3-alkylpyridinium alkaloids: (m) ref 1986, 51, 404-407.
(8) Mancini et al. prepared aldehyde 10 from bromide 9 using DMSO
(5) For glutaconaldehyde and its derivatives, including glutaconal-
as an oxidant: Mancini, I.; Guella, G.; Pietra, F. Helv. Chim. Acta 1991,
dehyde bisacetals, see: Becher, J. Synthesis 1980, 589-612. J. Org. Chem., Vol. 64, No. 10, 199911,10 using potassium tert-butoxide in the presence of 18- crown-6 as a base, afforded (Z)-alkene 12 nearly as a single geometrical isomer (E-isomer < 2%). Deprotection to alcohol 13 and subsequent treatment with TsCl/ pyridine led to tosylate 14 which was used immediately without purification for the next step to prevent poly- merization. Imine 1511 was deprotonated with LDA in THF at -78 °C, and the resulting lithio enamine was alkylated with tosylate 14. Hydrolysis of the imine functionality upon aqueous workup provided ketone 16 in 70% yield. Wittig reaction of 16 with the ylide generated from phosphonium salt 5 furnished glutacon- aldehyde precursor 17 as a 9:1 mixture of E/Z-isomers in 63% yield. In contrast to ketone 6 (Scheme 2), the use of an excess (4.7 equiv) of ylide and a modified temper- ature protocol were found essential to achieve this result.
chloride in refluxing ethanol. Tosylation, azide substitu-
In some experiments 17 was accompanied by minor
tion, and reduction afforded amine 4 which was isolated
amounts (<15%) of aldehyde 18 arising from dimethyl
as its dihydrochloride salt 4a. The overall yield of this
acetal hydrolysis which was of no consequence for the
10-step sequence starting from 19 was 4.3%.
subsequent reaction. This sequence allowed us to obtain
With both components in hand, the stage was set for
glutaconaldehyde precursor 17 in 27% overall yield in
the condensation of 17 and 4 to niphatoxin B. Glutacon-
six steps starting from 9 (Scheme 3).
aldehyde 3 (Scheme 1) was liberated by hydrolysis of the acetal functionalities, applying the conditions found in
For the construction of the amine component 4, propyn-
the model reaction with bisacetal 7, and cyclized in situ
1-ol was deprotonated with n-BuLi, and the resulting
with amine 4 to give niphatoxin B (2) in 45% yield after
dianion was alkylated with THP-protected bromo alcohol
19, giving the known propargyl alcohol 2012 which was
1H and 13C NMR data16 of synthetic niphatoxin B (2)
to provide bromide 21 (Scheme
were identical with those reported for the natural prod-
4). Generation of the enolate14 from ester 22, followed
uct.1 In the FAB-MS, synthetic 2 revealed a base peak
by addition of bromide 21, afforded alkylation product
at m/z 524 and prominent peaks at m/z 295 and 230
23 which was reduced with LiAlH4 to alcohol 24. Partial
which were assigned to [M+] and the fragments of C-N
hydrogenation using Lindlar catalyst and subsequent
cleavage, respectively. The dimethylation product of
Swern oxidation gave glutaraldehyde monoacetal 26.
synthetic niphatoxin B showed the same behavior in 1H
Glutaraldehyde-pyridine cyclization15 and THP depro-
and 13C NMR experiments17 as that of natural niphatox-
tection to pyridine alcohol 27 were achieved in one
laboratory step by treatment with hydroxylammonium
In conclusion, the first synthesis of niphatoxin B has
been accomplished. We have shown that the reaction of
(9) For oxidation with pyridine N-oxide: (a) Waugh, K. M.; Berlin,
primary amines with 2-substituted glutaconaldehydes,
K. D. J. Org. Chem. 1984, 49, 873-878. With trimethylamine N- oxide: (b) Godfrey, A. G.; Ganem, B. Tetrahedron Lett. 1990, 31, 4825-
generated in situ from the corresponding bisacetals,
provides a practical entry to 3-substituted pyridinium
(10) Prepared in one step from commercially available 3-(3-pyridyl)-
salts for which classical approaches, i.e., halide or sul-
1-propanol in 80% yield: Staab, H. A.; Zipplies, M. F.; Mu
Storch, M.; Krieger, C. Chem. Ber. 1994, 127, 1667-1680.
fonate displacement by pyridines, are not considered
(11) Cuvigny, T.; Normant, H. Synthesis 1977, 198-200.
convenient. Our protocol allows the synthesis of pyri-
(12) Vig et al. used LiNH2/liquid NH3 for this conversion: Vig, O.
P.; Sharma, M. L.; Kapur, J.; Thapar, S.; Gupta, R. Indian J. Chem. Sect. B 1990, 29, 606-610.
(16) Since our 1H and 13C NMR data of synthetic 2 matched exactly
(13) Harnden, M. R.; Jarvest, R. L. J. Chem. Soc., Perkin Trans. 1
those reported for the natural product when recorded in CD3OD, we
1988, 2777-2784
assume that also Talpir et al. used CD3OD as the solvent for their
(14) Cooke, M. P., Jr.; Gopal, D. J. Org. Chem. 1994, 59, 260-263.
NMR experiments and not CDCl3 as stated in ref 1. Especially the 1H
(15) Spitzner, D. In Houben-Weyl-Methoden der Organischen Che-
NMR chemical shifts of the pyridinium protons show strong solvent
mie, 4th ed.; Kreher, R. P., Ed.; Thieme: Stuttgart, 1992; Vol. E7b, pp
dependence. For details, see the Experimental Section.
(17) See the Supporting Information. J. Org. Chem., Vol. 64, No. 10, 1999
by using Merck Kieselgel 60 and Merck aluminum oxide 90 (70-230 mesh, activity II-III). Spots were visualized with ultravioletlight (254 nm) or detected by exposure to iodine fumes. Infraredspectra were recorded with an FT-IR spectrometer. 1H and 13CNMR spectra were obtained at 250 and 62 MHz, respectively. Compounds which were not submitted for or did not passelemental analysis were judged to be of >95% purity on the basisof TLC homogenity and 1H NMR analyses (see SupportingInformation). 1,1-Dimethoxy-4-(1,3-dioxolan-2-yl)-2-methylbut-2-ene (7) (E/Z-mixture). Phosphonium salt 5 (8.87 g, 20 mmol) was suspended in THF (60 mL) and cooled to -20 °C. n-BuLi (12.5 mL, 20 mmol, 1.6 M in hexane) was added dropwise under a nitrogen atmosphere. After 1 h at this temperature, ketone 6 (2.48 g, 21 mmol) in THF (10 mL) was added. The cooling bath was removed, and stirring was continued for 16 h. Water (300 mL) was added, the layers were separated, and the aqueous phase was extracted with ether. The combined organic layers were washed with water and brine, dried, and concentrated in vacuo. The residue was purified by bulb-to-bulb distillation (0.05 Torr, ot 50 °C) to afford 7 (2.2 g, 54%, 9:1 mixture of E/Z-isomers) as colorless oil. IR (film): 2890, 2830 cm-1. 1H NMR (CDCl3): δ 5.64 (m, 0.1 H) and 5.52 (m, 0.9 H), 4.93 (d, J ) 0.9 Hz, 1 H), 4.89 (t, J ) 4.8 Hz, 1 H), 3.80-4.05 (m, 4 H), 3.34 (s, 5.4 H) and 3.29 (s, 0.6 H), 2.49-2.57 (m, 2 H), 1.70-1.75 (m, 2.7 H), and 1.62-1.65 (m, 0.3 H). 13C NMR (CDCl3): δ 135.6 (major), 134.9 (minor), 123.5 (major), 122.5 (minor), 103.9, 102.4, 64.9 (2C), 54.0 (minor, 2C), 53.5 (major, 2C), 32.55 (minor), 32.38 (major), 17.9. N-Cyclohexyl-3-methylpyridinium Chloride (8). 7 (440
mg, 2.2 mmol) was dissolved in CH2Cl2 (4 mL), FeCl3/SiO2 catalyst7 (100 mg) was added, and the mixture was stirred for 5 min. Then cyclohexylamine hydrochloride (400 mg, 2.9 mmol) in methanol (0.5 mL) was added. After 1 h n-butanol (5 mL) and Et3N (300 mg, 3.0 mmol) were added and CH2Cl2 was removed in vacuo. The solution was refluxed for 16 h. Then the solvent was removed in vacuo, and water (5 mL), a concentrated Na2CO3 solution (5 mL), and ether (5 mL) were added. The layers were separated, and the aqueous layer was extracted with ether. The aqueous phase was concentrated in vacuo to dryness, and 8 was extracted from the residue with CH2Cl2. The crude product was further purified by column chromatography (SiO2, gradient CH2Cl2/methanol: 0-15% methanol) to afford 8 (180 mg, 39%) as a light brown oil. IR (film): 3039, 1630 cm-1. 1H NMR
dinium salts in only three steps from acyclic starting
(CDCl3): δ 9.71 (br s, 1 H), 9.52 (d, J ) 6.1 Hz, 1 H), 8.27 (br d,
materials18 by the sequence of (a) alkylation of 1,1-
J ) 7.8 Hz, 1 H), 8.12 (dd, J ) 7.8, 6.1 Hz, 1 H), 5.19 (tt, J )12.0, 4.0 Hz, 1 H), 2.70 (s, 3 H), 1.90-2.31 (m, 6 H), 1.31-1.83
dimethoxy-2-propanone via its azaenolate, (b) Wittig
(m, 4 H). 1H NMR (CD3OD): δ 9.00 (s, 1 H), 8.92 (d, J ) 5.9 Hz,
olefination with 2-(1,3-dioxolan-2-yl)ethyltriphenylphos-
1 H), 8.42 (d, J ) 7.9 Hz, 1 H), 7.93-8.06 (m, 1 H), 4.57-4.79
phonium bromide, and (c) cyclization with a primary
(m, 1 H), 2.60 (s, 3 H), 1.26-2.28 (m, 10 H). 13C NMR (CD3OD):
amine. It should be equally useful for making a range of
δ 147.4, 144.1, 141.6, 141.5, 128.9, 73.3, 34.5 (2C), 26.5 (2C),
analogues for biological investigations. 6-(Tetrahydro-2-pyranyloxy)hexanal (10). With Pyri- dine N-Oxide. A mixture of 1-bromo-6-(tetrahydro-2-pyranyl- Experimental Section
oxy)hexane (9) (16.3 g, 90 mmol), NaHCO3 (16.8 g, 200 mmol), and pyridine N-oxide (19.0 g, 200 mmol) in toluene (150 mL) General. Compounds 9 and 19 were prepared according to
was refluxed for 4 h, using a Dean-Stark water trap, to remove
literature procedures8,19,20 from the corresponding diols. Com-
the water formed in the reaction. After cooling, the mixture was
mercial reagent grade solvents and chemicals were used as
filtered and the solution was concentrated in vacuo. The crude
obtained except as indicated below. DMPU (absolute, puriss. over
product was purified by column chromatography (SiO
molecular sieve) was purchased from Fluka and DMSO (dried)
petroleum ether 8/2) to afford 10 (6.88 g, 38%) as a colorless oil.
from Merck. THF was distilled from sodium benzophenone ketyl.
Analytical data were in agreement with those in the literature.8
Pyridine and Et3N were stored over KOH pellets. Prior to use
With Trimethylamine N-Oxide (TMANO). 1-Bromo-6-
in Swern oxidation, CH2Cl2 was distilled from P2O5. Petroleum
(tetrahydro-2-pyranyloxy)hexane (9) (0.53 g, 2 mmol) was dis-
ether refers to the 40-60 °C boiling fraction. Solvents used for
solved in DMSO (4 mL). TMANO (0.60 g, 8 mmol) was added,
column chromatography were distilled prior to use. All metal-
and the mixture was stirred for 5 h. The mixture was poured
lorganic reactions were run in flame-dried glassware under
into a half-saturated NaCl solution and extracted with ether.
nitrogen. Organic extracts were dried over anhydrous Na2SO4.
The combined organic layers were washed with water and brine,
For thin-layer chromatography (TLC) analysis, precoated TLC
dried, and concentrated in vacuo. The residue was purified as
plates (Merck Kieselgel 60 F254 and Merck aluminum oxide 60
indicated above to give 10 (0.36 g, 90%) as a colorless oil.
F254 neutral) were used, and column chromatography was done
(Z)-3-[9-(Tetrahydro-2-pyranyloxy)non-3-en-1-yl]pyri- dine (12). To a stirred suspension of phosphonium salt10 11 (23.4
(18) For a recent example and references for the preparation of
g, 50.5 mmol) and 18-crown-6 (0.9 g, 3.4 mmol) in THF (60 mL)
pyridinium salts from acyclic starting materials, see: Yu, L.-B.; Chen,
was added a solution of potassium tert-butylate (8.5 g, 75.8
D.; Li, J.; Ramirez, J.; Wang, P. G. J. Org. Chem. 1997, 62, 208-211.
mmol) in THF (60 mL) dropwise under a nitrogen atmosphere
(19) Kang, S.-K.; Kim, W.-S.; Moon, B.-H. Synthesis 1985, 1161-
at 0 °C. After 30 min at 0 °C, the solution was cooled to -78 °C
(20) Chapman, O. L.; Mattes, K. C.; Sheridan, R. S.; Klun, J. A. J.
and aldehyde 10 (6.75 g, 33.7 mmol) in THF (30 mL) was added Am. Chem. Soc. 1978, 100, 4878-4884.
over a period of 30 min. After 30 min, the cooling bath was
J. Org. Chem., Vol. 64, No. 10, 1999
removed and the mixture was stirred for additional 2 h. The
J ) 7.8, 2.2, 1.7 Hz, 1 H), 7.20 (ddd, J ) 7.8, 4.8, 0.8 Hz, 1 H),
reaction was quenched with water (100 mL), the layers were
5.67 (t, J ) 7.1 Hz, 0.1 H), 5.48 (t, J ) 7.1 Hz, 0.9 H), 5.37 (m,
separated, and the aqueous phase was extracted with EtOAc.
2 H), 4.92 (s, 1 H), 5.03 (t, J ) 4.4 Hz, 0.9 H), 4.89 (t, J ) 4.4
The combined organic layers were washed with water and brine,
Hz, 0.1 H), 3.81-4.04 (m, 4 H), 3.33 (s, 5.4 H), 3.27 (s, 0.6 H),
dried, and concentrated in vacuo. The residue was purified by
2.66 (t, J ) 7.8 Hz, 2 H), 2.54 (m, 2 H), 2.35 (m, 2 H), 1.84-2.09
column chromatography (SiO2, ether) to afford 12 (7.3 g, 81%)
(m, 4 H), 1.19-1.49 (m, 7 H). 13C NMR (CDCl3): δ 149.6, 146.8,
as pale yellow oil. IR (film): 3006, 2938, 2860 cm-1. 1H NMR
139.5, 137.5, 136.3, 131.5, 127.6, 123.2, 122.7, 104.0, 103.2, 64.9
(CDCl3): δ 8.44 (br s, 1H), 8.42 (dd, J ) 4.9, 1.7 Hz, 1 H), 7.48
(2C), 54.2 (2C), 33.1, 32.5, 31.3, 29.5, 29.2, 29.0, 28.7 (2C), 27.3.
(ddd, J ) 7.7, 2.2, 1.7 Hz, 1 H), 7.18 (ddd, J ) 7.7, 4.8, 0.8 Hz,
11-(Tetrahydro-2-pyranyloxy)undec-2-yn-1-ol (20). In a
1 H), 5.38 (m, 2 H), 4.57 (m, 1 H), 3.80-3.92 (m, 1 H), 3.65-
three-necked flask with a mechanical stirrer, a solution of
3.78 (m, 1 H), 3.43-3.55 (m, 1 H), 3.30-3.42 (m, 1 H), 2.66 (t, J
2-propyn-1-ol (1.12 g, 20 mmol) in THF (75 mL) was cooled to
) 7.6 Hz, 2 H), 2.35 (dt, J ) 7.1, 7.0 Hz, 2 H), 1.20-2.02 (m, 14
-78 °C under a nitrogen atmosphere. n-BuLi (25 mL, 40 mmol,
H). 13C NMR (CDCl3): δ 150.1, 147.3, 137.2, 135.8, 131.2, 127.9,
1.6 M in hexane) was slowly added. After addition was complete,
123.1, 98.9, 67.5, 62.3, 33.0, 30.8, 29.6, 29.4, 28.7, 27.2, 25.9,
the temperature was allowed to rise to -30 °C. After 45 min at
this temperature, a solution of 19 (2.93 g, 10 mmol) in DMPU (Z)-9-(3-Pyridyl)non-6-en-1-ol (13). To a stirred solution of
(40 mL) and THF (30 mL) was added. The cooling bath was
12 (1.9 g, 6.3 mmol) in methanol (35 mL) was added TsOH (1.24
removed, and stirring was continued for an additional 16 h.
g, 6.5 mmol). Stirring was continued for 4 h. A saturated
Water (100 mL) was added, the layers were separated, and the
NaHCO3 solution (45 mL) and water (90 mL) were added, and
aqueous layer was extracted with EtOAc. The combined organic
the mixture was extracted with EtOAc. The combined organic
layers were washed with water and brine, dried, and concen-
phases were washed with a saturated NaHCO3 solution and
trated in vacuo. The residue was purified by column chroma-
brine, dried, and concentrated in vacuo. The residue was purified
tography (SiO2, petroleum ether/EtOAc 7/3) to give 20 (1.35 g,
by column chromatography (SiO2, CH2Cl2/methanol 9/1) to afford
47%) as a colorless oil. Analytical data were in agreement with
13 (1.16 g, 84%) as pale yellow oil. IR (film): 3315 cm-1. 1H NMR
(CDCl3): δ 8.44 (d, J ) 2.2 Hz, 1 H), 8.42 (dd, J ) 4.8, 1.7 Hz,
1-Bromo-11-(tetrahydro-2-pyranyloxy)undec-2-yne (21).
1 H), 7.49 (ddd, J ) 7.7, 2.2, 1.7 Hz, 1 H), 7.19 (ddd, J ) 7.7,
A stirred solution of 20 (7.0 g, 26.1 mmol) and CBr4 (13.0 g, 39.1
4.8, 0.8 Hz, 1 H), 5.29-5.47 (m, 2 H), 3.61 (t, J ) 6.6 Hz, 2 H),
mmol) in DMF (90 mL) was cooled to 0 °C. PPh3 (10.3 g, 39.1
2.67 (t, J ) 7.5 Hz, 2 H), 2.36 (dt, J ) 7.5. 6.7 Hz, 2 H), 1.83-
mmol) was added in one portion, and the mixture was stirred
2.13 (m, 3 H), 1.44-1.62 (m, 2 H), 1.17-1.37 (m, 4 H). 13C NMR
for 25 min at 0 °C. A half-saturated NaHCO3 solution (80 mL)
(CDCl3): δ 149.9, 147.2, 137.2, 136.1, 131.2, 127.9, 123.2, 62.6,
was added, and the mixture was extracted with petroleum ether.
33.0, 32.7, 29.3, 28.6, 27.1, 25.4. Anal. Calcd for C14H21NO: C,
The combined organic layers were washed with water and brine,
76.67; H, 9.65; N, 6.39. Found: C, 76.23; H, 9.65; N, 6.39.
dried, and concentrated in vacuo. The residue was purified by
(Z)-1,1-Dimethoxy-12-(3-pyridyl)dodec-9-en-2-one (16).
column chromatography (SiO2, petroleum ether/EtOAc 9/1) to
Alcohol 13 (1.06 g, 5.0 mmol) was dissolved in pyridine (15 mL)
give 21 (7.90 g, 91%) as a colorless oil. IR (film): 2312, 2234
and cooled to -10 °C, and p-toluenesulfonyl chloride (1.05 g, 5.5
cm-1. 1H NMR (CDCl3): δ 4.53-4.62 (m, 1 H), 3.93 (t, J ) 2.4
mmol) was added. After 15 min, the cooling bath was removed
Hz, 2 H), 3.81-3.98 (m, 1 H), 3.67-3.79 (m, 1 H), 3.44-3.56
and the mixture stirred for 2 h at room temperature. Water (20
(m, 1 H), 3.32-3.44 (m, 1 H), 2.16-2.29 (m, 2 H), 1.20-1.93 (m,
mL) and a saturated NaHCO3 solution (2 mL) were added, and
18 H). 13C NMR (CDCl3): δ 98.8, 88.3, 75.3, 67.6, 62.3, 30.8,
the solution was extracted with ether. The combined organic
29.7, 29.3, 29.0, 28.7, 28.3, 26.2, 25.5, 19.7, 18.9, 15.6.
layers were washed with water and brine, dried, and concen-
Methyl 2-(3,3-Dimethoxypropyl)-13-(tetrahydro-2-pyra-
trated in vacuo. The unstable product 14 was used immediately nyloxy)tridec-4-ynoate (23). A solution of diisopropylamine
for the following reaction without further purification.
(4.8 g, 47.0 mmol) in THF (100 mL) was cooled to -78 °C, and
A solution of diisopropylamine (3.04 g, 30 mmol) in THF (50
n-BuLi (25 mL, 40 mmol, 1.6 M in hexane) was added slowly.
mL) was cooled to -78 °C, and n-BuLi (15.6 mL, 25 mmol, 1.6
After 30 min at -78 °C, 22 (4.46 g, 25.3 mmol) in THF (20 mL)
M in hexane) was added dropwise. After 30 min at -78 °C,
was added dropwise and the solution was stirred for additional
imine11 15 (4.98 g, 25 mmol) in DMPU (5 mL) was added, and
30 min. 21 in DMPU (50 mL) was added, and after 1 h the
the solution was stirred for 1 h at -78 °C. Then tosylate 14 (5.0
cooling bath was removed. After 16 h water (100 mL) was added,
mmol, crude) in THF (15 mL) was added. After 2 h, the cooling
the layers were separated, and the aqueous layer was extracted
bath was removed and stirring was continued for 16 h at room
with EtOAc. The combined organic layers were washed with
temperature. The reaction was quenched with water (50 mL),
water and brine, dried, and concentrated in vacuo. The residue
the layers were separated, and the aqueous phase was extracted
was purified by column chromatography (SiO
with EtOAc. The combined organic phases were washed with
EtOAc 8/2) to give 23 (5.14 g, 47%) as a colorless oil. IR (film):
water and brine, dried, and concentrated in vacuo. The residue
was purified by column chromatography (SiO
3): δ 4.53-4.62 (m, 1 H), 4.32-4.41
(m, 1 H), 3.81-3.94 (m, 1 H), 3.65-3.79 (m, 1 H), 3.69 (s, 2.7 H,
16 (1.12 g, 70% from 13) as a pale yellow oil. IR (film): 1728
major diastereomer) and 3.67 (s, 0.3 H, minor diastereomer),
cm-1. 1H NMR (CDCl3): δ 8.44 (br d, J ) 2.2 Hz, 1 H), 8.42 (dd,
3.44-3.56 (m, 1 H), 3.26-3.44 (m, 1 H), 3.32 (minor diastere-
J ) 4.8, 1.7 Hz, 1 H), 7.49 (ddd, J ) 7.8, 2.2, 1.7 Hz, 1 H), 7.19
omer, s, 0.6 H) and 3.30 (major diastereomer, d, J ) 2.0 Hz, 5.4
(ddd, J ) 7.8, 4.8, 0.8 Hz, 1 H), 5.37 (m, 2 H), 4.45 (s, 1 H), 3.40
H), 2.49-2.61 (m, 1 H), 2.29-2.47 (m, 2 H), 2.07-2.16 (m, 2 H),
(s, 6 H), 2.65 (t, J ) 7.6 Hz, 2 H), 2.53 (t, J ) 7.3 Hz, 2 H), 2.34
(m, 2 H), 1.91 (m, 2 H), 1.55 (m, 2 H), 1.24 (br s, 6 H).13C NMR
67.6, 62.3, 52.8, 52.5, 51.6, 44.7, 33.7, 31.9, 30.8, 30.0, 29.7, 29.3,
(CDCl3): δ 205.6, 150.1, 147.3, 137.2, 135.8, 131.2, 127.8, 123.1,
29.1, 28.9, 28.7, 26.2 (2C), 25.5, 21.7, 19.7, 18.7.
104.3, 54.7 (2 C), 37.2, 33.0, 29.3, 29.0, 28.9, 28.7, 27.1, 22.9. 2-(3,3-Dimethoxypropyl)-13-(tetrahydro-2-pyranyloxy)- (3Z)-3-[11-Dimethoxymethyl-13-(1,3-dioxolan-2-yl)tridec- tridec-4-yn-1-ol (24). A stirred suspension of LiAlH 3,11-dien-1-yl]pyridine (17). Phosphonium salt 5 (7.27 g, 16.4
23.4 mmol) in THF (65 mL) was cooled to 0 °C. Ester 23 (5.01 g,
mmol) was suspended in THF (100 mL) and cooled to -20 °C.
11.74 mmol) in THF (30 mL) was added slowly at this temper-
n-BuLi (11.0 mL, 17.6 mmol, 1.6 M in hexane) was added
ature, and the mixture was stirred for an additional 3 h at room
dropwise, and the solution was stirred for 1 h at -20 °C. Then
temperature. Then water (32.5 mL) was added dropwise to
the reaction was cooled to -78 °C and ketone 16 (1.12 g, 3.51
quench the reaction upon which a white solid precipitated. The
mmol) in THF (15 mL) was added dropwise. The solution was
solution was decanted, and the solid was extracted with ether
allowed to reach room temperature over a period of 16 h. Water
(3 × 100 mL). The combined organic phases were washed with
(150 mL) was added, the layers were separated, and the aqueous
water and brine, dried, and concentrated in vacuo. The residue
phase was extracted with EtOAc. The combined organic layers
was purified by column chromatography (SiO
were washed with water and brine, dried, and concentrated in
EtOAc 1/1) to afford 24 (3.42 g, 73%) as pale yellow oil. IR
vacuo. The residue was purified by column chromatography (SiO2, ether) to afford 17 (0.88 g, 63%, E/Z-mixture 9:1) as pale yellow oil. IR (film): 2929, 2857 cm-1. 1H NMR (CDCl3): δ 8.44
(21) Poulain, S.; Noiret, N.; Nugier-Chauvin, C.; Patin, H. Liebigs
(br d, J ) 2.2 Hz, 1 H), 8.43 (dd, J ) 4.8, 1.7 Hz, 1 H), 7.50 (ddd,
Ann./Recl. 1997, 35-40. J. Org. Chem., Vol. 64, No. 10, 1999
(film): 3450, 2362 cm-1. 1H NMR (CDCl3): δ 4.54-4.60 (m, 1
(200 mL) and extracted with ether. The combined organic phases
H), 4.35 (t, J ) 5.1 Hz, 1 H), 3.81-3.93 (m, 1 H), 3.58-3.79 (m,
were washed with water and brine, dried, and evaporated in
1 H), 3.64 (d, 2 H), 3.26-3.56 (m, 2 H), 3.32 (s, 6 H), 2.20-2.31
vacuo. The residue was immediately dissolved in DMF (20 mL),
(m, 2 H), 2.07-2.20 (m, 2 H), 1.20-1.91 (m, 23 H). 13C NMR
NaN3 (1.5 g, 23.1 mmol) was added, and the solution was stirred
(CDCl3): δ 104.7, 98.8, 67.6, 62.3, 55.5, 54.5, 35.1, 34.3, 30.8,
for 16 h at 70 °C. Then water (200 mL) was added, and the
29.7, 29.6, 29.3, 29.1, 29.0 (2C), 28.8, 26.2, 26.1, 25.5, 24.2, 22.1,
solution was extracted with petroleum ether. The combined
19.7, 18.7. Anal. Calcd for C23H42O5: C, 69.31; H, 10.62. Found:
organic layers were washed with water and brine, dried, and
concentrated in vacuo. The residue was purified by column
(Z)-2-(3,3-Dimethoxypropyl)-13-(tetrahydro-2-pyranyl-
chromatography (SiO2, EtOAc) to afford 29 (470 mg, 73%) as a oxy)tridec-4-en-1-ol (25). Methanol (50 mL), quinoline (200
colorless oil. IR (film): 2095 cm-1. 1H NMR (CDCl3): δ 8.40 (dd,
µL), Lindlar catalyst (250 mg, Fluka), and 24 (500 mg, 1.25 J ) 2.3, 0.7 Hz, 1 H), 8.37 (dd, J ) 4.8, 1.7 Hz, 1 H), 7.42 (ddd,
mmol) were placed in a hydrogenation flask and hydrogenated
J ) 7.8, 2.3, 1.7 Hz, 1 H), 7.13 (ddd, J ) 7.8, 4.8, 0.7 Hz, 1 H),
for 15 min at atmospheric pressure. The mixture was filtered,
5.37-5.58 (m, 2 H), 3.19 (d, J ) 5.9 Hz, 2 H), 3.33 (t, J ) 6.9
and the filtrate was concentrated in vacuo to afford 25 as a pale
Hz, 2 H), 2.01-2.15 (m, 2 H), 1.42-1.61 (m, 2 H), 1.17-1.42
yellow oil in quantitative yield. The crude product was used for
(m, 10 H). 13C NMR (CDCl3): δ 149.9, 147.3, 136.4, 135.6, 131.9,
the subsequent reaction without further purification. An ana-
126.6, 123.2, 51.4, 30.7, 29.4, 29.3, 29.1, 29.0, 28.8, 27.2, 26.6.
lytical sample was prepared by column chromatography (Al2O3,
(Z)-11-(3-Pyridyl)undec-9-en-1-amine Dihydrochloride
petroleum ether/ether gradient (0% ether-100% ether). IR
(4a). PPh3 (670 mg, 2.54 mmol) was added to a solution of azide
(film): 3454 cm-1. 1H NMR (CDCl3): δ 5.29-5.53 (m, 2 H), 4.53-
29 (470 mg, 1.72 mmol) in pyridine (1.6 mL) at 0 °C and was
4.63 (m, 1 H), 4.33 (t, J ) 5.1 Hz, 1 H), 3.81-3.94 (m, 1 H),
stirred for 24 h at room temperature. The solution was then
3.65-3.79 (m, 1 H), 3.22-3.63 (m, 4 H), 3.30 (s, 6 H), 1.20-2.24
cooled to 0 °C, concentrated NH3 (430 µL) was added, and the
(m, 27 H). 13C NMR (CDCl3): δ 131.6, 126.5, 105.0, 98.9, 67.7,
solution was stirred for another 24 h at room temperature.
65.4, 62.4, 52.8, 41.0, 32.6, 30.8, 30.0, 29.8, 29.6, 29.43, 29.41,
Pyridine was evaporated and the residue mixed with 2 N HCl
29.2, 29.0, 27.3, 26.2, 25.7, 25.5, 19.7. Anal. Calcd for C23H44O5:
(10.2 mL). The mixture was extracted with ether (4 × 15 mL).
C, 68.96; H, 11.07. Found: C, 68.83; H, 11.09.
The aqueous phase was concentrated in vacuo to afford 4a (517 (Z)-2-(3,3-Dimethoxypropyl)-13-(tetrahydro-2-pyranyl-
mg, 94%) as a pale yellow oil. IR (film): 3384 cm-1. 1H NMR
oxy)tridec-4-en-1-al (26). A solution of oxalyl chloride (228 mg,
(CD3OD): δ 8.73 (br s, 2 H), 8.52 (br d, J ) 8.3 Hz, 1 H), 8.05
1.8 mmol) in dry CH2Cl2 (2 mL) was cooled to -78 °C, and a
(dd, J ) 8.3, 6.1 Hz, 1 H), 5.54-5.78 (m, 2 H), 3.70 (br d, J )
solution of DMSO (281 mg, 3.6 mmol) in dry CH2Cl2 (0.5 mL)
7.1 Hz, 2 H), 2.91 (t, J ) 7.6 Hz, 2 H), 2.12-2.25 (m, 2 H), 1.60-
was added dropwise under a nitrogen atmosphere. After 30 min
1.69 (m, 2 H), 1.29-1.37 (m, 10 H). 13C NMR (CD
alcohol 25 (600 mg, 1.5 mmol) in dry CH
142.8, 141.9, 140.4, 135.4, 128.5, 125.4, 40.9, 31.1, 30.5, 30.3,
slowly and the mixture was stirred for an additional 30 min.
Et3N (1.05 mL) was added, and the cooling bath was removed. After the reaction mixture reached room temperature, water (3
Niphatoxin B (2). 17 (72 mg, 0.18 mmol) was dissolved in
mL) was added and the solution was extracted with CH
CH2Cl2 (2 mL), FeCl3/SiO2 catalyst (30 mg) was added, and the
combined organic layers were washed with water and brine,
mixture was stirred for 5 min. Dihydrochloride 4a (83 mg, 0.26
dried, and concentrated in vacuo. The residue was purified by
mmol) in methanol (0.6 mL) was added, and stirring was
continued for 30 min. n-BuOH (3 mL) was added, and CH2Cl2
give 26 (480 mg, 80% from 24) as a pale yellow oil. IR (film):
was removed in vacuo. Et3N (100 µL) was added, and the
solution was refluxed for 16 h. The solvents were evaporated in
3): δ 9.61 (d, J ) 2.0 Hz, 1 H), 5.39-
5.55 (m, 1 H), 5.23-5.37 (m, 1 H), 4.53-4.62 (m, 1 H), 4.34 (t, J
vacuo, and the residue was purified by column chromatography
) 5.3 Hz, 1 H), 3.81-3.94 (m, 1 H), 3.66-3.79 (m, 1 H), 3.44-
(SiO2, gradient CH2Cl2/methanol: 0-10% methanol) to afford
3.56 (m, 1 H), 3.33-3.44 (m, 1 H), 3.31 (s, 6 H), 2.15-2.46 (m,
2 (45 mg, 45%) as a light brown oil. IR (film): 3010, 2929, 2856,
3 H), 1.93-2.09 (m, 2 H), 1.21-1.87 (m, 22 H).13C NMR
1632, 1592, 1576, 1507, 1478, 1466, 1424, 1328, 1241, 1192,
1160, 1104, 1044, 1028, 834, 799, 718, 695, 523 cm-1.1H NMR
3): δ 204.3, 132.6, 125.2, 104.4, 98.8, 67.6, 62.3, 52.9, 52.8,
51.6, 30.8, 30.0, 29.9, 29.5, 29.42, 29.39, 29.2, 27.3, 26.7, 26.2,
(CD3OD): δ 8.94 (br s, 1 H), 8.85 (br d, J ) 6.0 Hz, 1 H), 8.46
(br d, J ) 8.2 Hz, 1 H), 8.30-8.41 (m, 4 H), 8.02 (dd, J ) 7.9,
6.1 Hz, 1 H), 7.63-7.73 (m, 2 H), 7.31-7.39 (m, 2 H), 5.47-5.63
(Z)-11-(3-Pyridyl)undec-9-en-1-ol (27). NH
(m, 2 H), 5.30-5.44 (m, 2 H), 4.61 (t, J ) 7.5 Hz, 2 H), 3.40-
mg, 11.0 mmol) was added to a solution of aldehyde 26 (880 mg,
3.47 (m, 2 H), 2.87 (t, J ) 7.8 Hz, 2 H), 2.70 (t, J ) 7.2 Hz, 2 H),
2.21 mmol) in 99% EtOH (20 mL), and the mixture was refluxed
2.31-2.43 (m, 2 H), 2.12-2.23 (m, 2 H), 1.84-2.08 (m, 4 H),
for 60 min. After the red solution reached room temperature,
1.61-1.77 (m, 2 H), 1.13-1.48 (m, 16 H). 1H NMR (CDCl3): δ
water (70 mL) and ether (70 mL) were added and the solution
9.43 (d, J ) 5.7 Hz, 1 H), 9.23 (s, 1 H), 8.43 (br s, 4 H), 8.21 (d,
was basified with 2 N NaOH. The layers were separated, and
J ) 7.8 Hz, 1 H), 7.97-8.10 (m, 1 H), 7.46-7.55 (m, 2 H), 7.16-
the aqueous layer was extracted with ether. The combined
7.27 (m, 2 H), 5.45-5.61 (m, 2 H), 5.27-5.45 (m, 2 H), 5.00 (t, J
organic layers were washed with water and brine, dried, and
7.1 Hz, 2 H), 3.39 (d, J ) 6.0 Hz, 2 H), 2.87 (t, J ) 7.2 Hz, 2
concentrated in vacuo. The residue was purified by column
H), 2.67 (t, J ) 7.2 Hz, 2 H), 1.11-2.53 (m, 26 H). 13C NMR
2, EtOAc) to afford 27 (290 mg, 54%) as a δ 150.26, 149.97, 147.65, 147.56, 146.67, 145.83,
colorless oil. IR (film): 3331 cm-1. 1H NMR (CDCl
145.25, 143.37, 139.57, 138.97, 138.48, 138.13, 133.10, 132.20,
J ) 2.3, 0.8 Hz, 1 H), 8.40 (dd, J ) 4.8, 1.7 Hz, 1 H), 7.48 (ddd,
129.05 (2 C), 127.81, 125.20, 125.05, 63.02, 33.79, 33.55, 32.47,
J ) 7.8, 2.3, 1.7 Hz, 1 H), 7.19 (ddd, J ) 7.8, 4.8, 0.8 Hz, 1 H),
31.46, 31.45, 30.57, 30.49, 30.33, 30.17, 30.03, 29.99, 29.94, 29.74,
5.44-5.64 (m, 2 H), 3.61 (t, J ) 6.6 Hz, 2 H), 3.37 (d, J ) 6.1
28.17, 28.07, 27.17. FAB MS, m/z (%): 524 (100) [M+], 431 (9),
Hz, 2 H), 2.49 (br s, 1 H), 2.06-2.21 (m, 2 H), 1.16-1.63 (m, 12
391 (7), 349 (7), 335 (15), 295 (15), 230 (15), 160 (8), 146 (13),
3): δ 149.7, 147.2, 136.6, 135.8, 132.1, 126.5,
123.3, 62.8, 32.8, 30.7, 29.5, 29.4, 29.3, 29.1, 27.2, 25.7. (Z)-3-(11-Azidoundec-2-en-1-yl)pyridine (29). A solution Supporting Information Available: 1H NMR spectra for
of p-toluenesulfonyl chloride (2.0 g, 10.4 mmol) in pyridine (10
compounds 2, 4a, 7, 8, 12, 16, 17, 21, 23, 27, and 29 and for
mL) was cooled to 0 °C, and alcohol 27 (580 mg, 2.36 mmol) in
the dimethylation product of 2. This material is available free
pyridine (3.4 mL) was added dropwise. The solution was stirred
of charge via the Internet at http://pubs.acs.org.
for 1 h at room temperature and cooled to 0 °C, and water (2mL) was added. After 5 min the solution was diluted with water
Commercialization of Biomarkers BULLETIN High-throughput genomics and proteomics technologies have led to the emergence and rapid proliferation of clinical use and commercial demand for biomarkers — BIOMARKERS: molecular indicators directly and highly predictive of a biological process or that have DEFINITIONS utility as theranostics to guide therapeutic decision making. These
INFORMAZIONI PERSONALI Nome REGINE FEDERICO Indirizzo UNITÀ OPERATIVA SEMPLICE DIPARTIMENTALE DI PATOLOGIE RETINICHE POLICLINICO DI TOR VERGATA VIALE OXFORD 00133 - ROMA Telefono +39 0620903572 Fax +39 0620903571 E-mail federico.regine@gmail.com ESPERIENZA LAVORATIVA - Ha espletato il Servizio Militare in qualità di S.Ten. Med. c.p.l. presso il Centro Addes
 J. Org. Chem. 1999, 64, 3778-3782
J. Org. Chem. 1999, 64, 3778-3782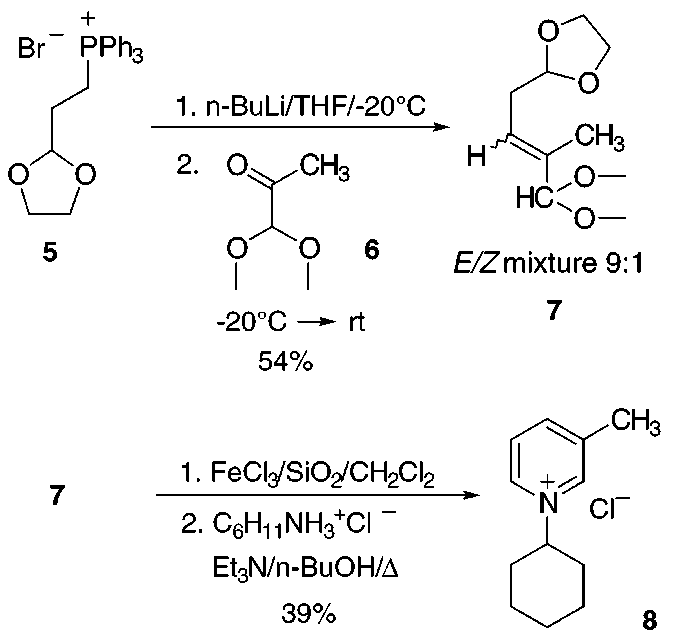
 J. Org. Chem., Vol. 64, No. 10, 1999
11,10 using potassium tert-butoxide in the presence of 18-
J. Org. Chem., Vol. 64, No. 10, 1999
11,10 using potassium tert-butoxide in the presence of 18-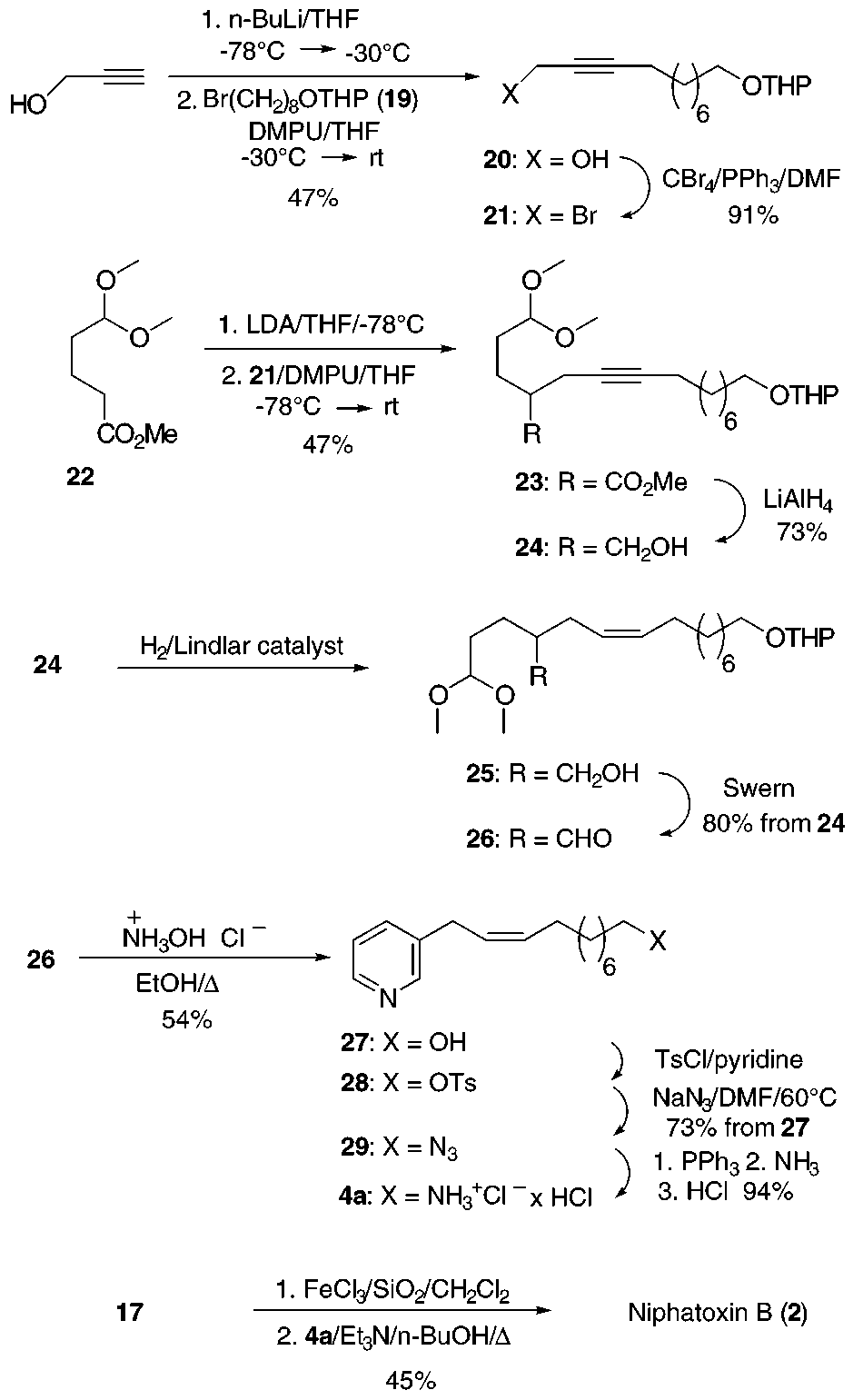 J. Org. Chem., Vol. 64, No. 10, 1999
by using Merck Kieselgel 60 and Merck aluminum oxide 90 (70-230 mesh, activity II-III). Spots were visualized with ultravioletlight (254 nm) or detected by exposure to iodine fumes. Infraredspectra were recorded with an FT-IR spectrometer. 1H and 13CNMR spectra were obtained at 250 and 62 MHz, respectively.
J. Org. Chem., Vol. 64, No. 10, 1999
by using Merck Kieselgel 60 and Merck aluminum oxide 90 (70-230 mesh, activity II-III). Spots were visualized with ultravioletlight (254 nm) or detected by exposure to iodine fumes. Infraredspectra were recorded with an FT-IR spectrometer. 1H and 13CNMR spectra were obtained at 250 and 62 MHz, respectively.