La tétracycline, connue sous le nom commercial Sumycin, agit en bloquant la fixation de l’ARNt sur la sous-unité 30S ribosomale, interrompant l’élongation de la chaîne protéique bactérienne. Ce mécanisme confère une activité sur un spectre large, incluant bactéries Gram positives, Gram négatives, rickettsies et spirochètes. Sa biodisponibilité digestive varie selon la prise alimentaire et les interactions avec les ions divalents comme calcium et magnésium. Sa diffusion tissulaire est importante, notamment dans les voies respiratoires et génito-urinaires. L’élimination se fait par voie rénale et biliaire. Les effets indésirables incluent photosensibilisation, troubles digestifs et coloration dentaire en cas d’administration précoce. Les guides thérapeutiques mentionnent sumycin prix, en soulignant la nécessité de restreindre son utilisation afin de limiter les résistances acquises.
Doi:10.1016/j.cardiores.2006.03.004
Cardiovascular Research 71 (2006) 30 – 39
C-reactive protein in atherosclerosis: A causal factor?
aHemostasis and Thrombosis Research Centre, Dept. of Hematology, Leiden University Medical Centre, Leiden, The Netherlands
bDepartment of Hematology, Room Ee 13.93, Erasmus University Medical Center, Dr. Molewaterplein 50, 3015 GE Rotterdam, The Netherlands
Received 3 August 2005; received in revised form 3 March 2006; accepted 6 March 2006
Atherosclerosis is considered a to be multifactorial disease driven by inflammatory reactions. The process of inflammation also
contributes to the pathogenesis of acute atherothrombotic events. C-reactive protein (CRP) is an acute phase protein and its concentration inserum reflects the inflammatory condition of the patient. Levels of CRP are consistently associated with cardiovascular disease (CVD) andpredict myocardial infarctions and stroke. Since CRP is present in the atherosclerotic lesion, it may actively contribute to the progression and/or instability of the atherosclerotic plaque. The role of CRP in inflammation and its causality in atherosclerosis are the subject of manyinvestigations but are not yet fully elucidated. This review focuses on recently identified mechanisms by which CRP may modulate andevolve the process of atherosclerosis. We discuss the function of CRP and review the most recent evidence for an independent role of CRP inthe development of atherosclerosis. Many studies suggest such a role, but a number of the described effects may be the result ofcontamination of the CRP preparations. D 2006 European Society of Cardiology. Published by Elsevier B.V. All rights reserved.
Keywords: C-reactive protein; Azide; Atherosclerosis; Inflammation; Thrombosis
predictive value in the pathogenesis of CVD. Many clinicaland population studies, with cross-sectional and nested
Inflammation plays a key role in the pathogenesis of
case – control designs, proved these inflammatory mediators
cardiovascular disease (CVD), acute atherothrombotic
events and atherosclerosis Inflammation also regu-
Several reports describe human CRP to be involved in
lates the production of the acute phase proteins such as C-
the induction of ischemic tissue damage in the brain,
reactive protein (CRP), fibrinogen and serum amyloid A
myocardial infarction and the increase of stroke volume in
The serum concentration of CRP can increase > 1000-
Most clinical studies report that CRP is an independent
fold upon inflammation and, with a half life of 19 h, CRP is
predictor of risk of atherosclerosis cardiovascular
a very stable downstream marker of the inflammatory
process Because CRP is such a sensitive indicator of the
myocardial infarction even after considering other
inflammatory process, it has been extensively studied
cardiovascular risk factors such as age, smoking, obesity,
whether plasma concentrations of CRP and other circulating
diabetes, hypercholesterolemia and hypertension. However,
inflammatory proteins (e.g. fibrinogen, interleukin-6) have a
absence of a relationship between CRP and risk ofmyocardial infarction has been reported as well, especiallyafter comprehensive adjustment for established risk factors
An early indication that CRP may be more than just a
* Corresponding author. Tel.: +31 10 4089448; fax: +31 10 4089470.
risk marker was the observation that, of several inflamma-
0008-6363/$ - see front matter D 2006 European Society of Cardiology. Published by Elsevier B.V. All rights reserved.
E. Paffen, M.P.M. deMaat / Cardiovascular Research 71 (2006) 30 – 39
tory markers studied (such as P-selectin, interleukin-6,
the development, instability and eventual rupture of the
interleukin-1, tumor necrosis factor-a (TNFa), soluble
intercellular adhesion molecule-1 (sICAM-1), fibrinogen),CRP emerged as the most powerful inflammatory predictorof future cardiovascular risk
Assuming that, in the physiological condition of human
pathology and the atherosclerotic lesion, CRP exerts direct
CRP is one of the substances present in the atheroscle-
functions, the question is whether it is possible to
rotic lesion, more specifically in the vascular intima, where
discriminate between the direct effects of CRP and the
it co-localizes with monocytes, monocyte-derived macro-
parallel presence of other factors that determine the risk of
phages and lipoproteins This localization makes a
CVD. It was recently observed that several of the
direct contribution to the atherosclerotic process possible.
biological effects contributed to CRP in vitro are in fact
CRP is a phylogenetically highly conserved plasma
caused by contamination of the CRP preparation by azide
protein, with homologues in vertebrates and many inverte-
or bacterial lipopolysaccharides However, in other
brates, that is part of the systemic response to inflammation
studies a contribution of contamination can be excluded,
It is an acute phase protein and a member of the family
indicating that a causal role of CRP is clearly present.
of pentraxins. CRP was originally observed in 1930 in the
Examples are the recent ex vivo and in vivo studies
plasma of patients with acute infections, where it reacted
where CRP was administered to healthy volunteers
with the C polysaccharide of pneumococcus
and the in vitro experiments on the effect of CRP on
The major part of the CRP present in the plasma comes
tissue plasminogen activator activity, interleukin-1h and
from the liver, where the synthesis of CRP is mainly
tumor necrosis factor-a in human aortic endothelial cells
regulated by interleukin-6, which in turn is upregulated by
The functional effects of CRP that may be relevant
other inflammatory cytokines such as interleukin-1 and
for the development of CVD and will be discussed in this
tumor necrosis factor-a Small amounts of CRP can
also be produced locally For example, CRP has beendetected on the surface of about 4% of normal bloodlymphocytes and it has been demonstrated that this CRP is
produced by the lymphocytes themselves CRP alsocan be produced locally in atherosclerotic lesions by SMCs
The first step in the development of an atherosclerotic
plaque and the resulting local inflammatory process is
The structure of CRP is important for its stability and for
endothelial dysfunction. Upon injury, endothelial cells
the execution of its function CRP is composed of
(ECs) express the vascular cell adhesion molecule-1
five identical, 21,500 Da subunits. Upon dissociation of its
(VCAM-1), intracellular adhesion molecule-1 (ICAM-1)
pentameric structure, CRP subunits undergo a spontaneous
and the endothelial leukocyte adhesion molecule-1
and irreversible conformational change. The loss of the
(ELAM) on the cell surface Leukocytes, especially
pentameric structure of CRP results in modified or
T-lymphocytes (CD8+ cytotoxic T-cells, CD4+ helper Th1/
monomeric CRP (mCRP), which is a naturally occurring
Th2-cells) and monocytes, are then recruited from the
form of CRP and it is a tissue-based rather than a serum-
blood and cross the endothelial cell barrier via a process
based molecule mCRP is less soluble than CRP and
called diapedesis. Lipoprotein particles (LDLs and var-
tends to aggregate, and it has been described to induce
iants such as oxidized LDL) accumulate in the lesion, are
mRNA of chemokines and the expression of adhesion
taken up by monocyte-derived macrophages, which as a
molecules in human cultured coronary artery endothelial
result develop into foam cells, and form a fatty streak.
cells (HCAECs) Thus, next to circulating native
Smooth muscle cells (SMCs) proliferate and extracellular
pentameric CRP, mCRP can also promote a pro-inflamma-
matrix components extend to form a fibrous cap,
tory phenotype and exert atherogenic effects in human
enclosing and defining the morphology of the atheroscle-
endothelial cells, although it may be in less potent manner
than native CRP In ApoE(À/À) mice, mCRP has been
The cells involved in formation of the atherosclerotic
described to have opposite effects on atherosclerosis
plaque (e.g. ECs, monocytes, T-cells, SMCs) are stimulated
compared to normal CRP These data may explain in
to produce many different substances, such as inflamma-
part the conflicting activities previously reported for CRP in
tory mediators (interleukin-6, tumor necrosis factor-a,
interleukin-1) complement factors (C1q, C3, C5 –C9) chemokines (monocyte chemoattractant pro-tein-1, interleukin-8), adhesive molecules (selectins P/E,
4. Functionality of CRP and purity of CRP preparations
integrins CD18/CD11) metalloproteinases (MMP-1/9)collagenases, reactive oxygen species (such as nitric
The assumption that CRP is a causal factor in the
oxide (NO)) and CRP These mediators contribute to
development of the atherosclerotic lesion is based on its
E. Paffen, M.P.M. deMaat / Cardiovascular Research 71 (2006) 30 – 39
rapid accessibility to the plaque, localization in the plaque
rosis. Therefore, in this review, we will discuss the possible
and the results of in vitro studies, in which CRP has been
direct roles of CRP in atherosclerosis.
demonstrated to actively contribute to inflammatory pro-cesses.
The involvement of CRP in several mechanisms execut-
ing and maintaining the inflammatory process has beenwidely studied with the use of different commercially
Inflammatory mechanisms play a central role in all
human CRP preparations. These CRP preparations differ
phases of atherosclerosis, from the initial recruitment of
in source (serum, plasma, recombinant) and quality.
circulating leukocytes to the arterial wall to the rupture of
Use of commercially CRP preparations introduces two
unstable plaques, which results in the clinical manifestations
means of interference: 1) by contamination with bacterial
of the disease. CRP may be involved in each of these stages
lipopolysaccharides (LPS) or 2) by contamination with the
by direct influencing processes like complement activation,
apoptosis, vascular cell activation, monocyte recruitment,
Presence of endotoxin in commercially available CRP
lipid accumulation and thrombosis. Each of these processes
preparations has been eloquently described and we
offers several mechanisms by which CRP may influence its
ourselves measured concentrations between 3 and 600 pg
progress, as will be described in more detail below.
endotoxin/100 Ag CRP in different purified and recombinantCRP preparations (with the Limulus Amebocyte test which
is endotoxin-positive > 10 pg = 0.12 EU endotoxin present;unpublished data 2003). The presence of even very low
Activation of the classical pathway of the complement
concentrations of endotoxin can interact with the contents of
system is a well known and direct biological function of
the vessel wall activate gene expression and a cascade
CRP Via this action, CRP directly amplifies and
of reactions in monocytes activate procoagulant activity
facilitates innate immunity a process that has
in monocytes and macrophages through induction of
already been associated with initiation and progression of
tissue factor Endotoxins trigger an atherogenic re-
CVD for a long time. In situ hybridization showed intense
sponse in SMCs and block the induction of secretion of
mRNA signals for CRP and complement component C4 in
interleukin-1 and MCP-1 by human endothelial cells
SMCs and macrophages present in the thickened intima of
The amount of sodium azide present in different
the lesion. CRP also co-localizes with C5 – C9, the
commercially CRP preparations, varies from 0.05% to
membrane attack complex, of complement Activation
1.00% of the CRP present. Recent studies describe an
of this membrane attack complex (MAC) is initiated by the
effect of this preservative, when tested in vitro, on several
direct binding of CRP to C1q, also present in the
processes involved in atherosclerotic development, which
atherosclerotic lesion and characterized by elevated
were formerly subscribed to CRP. Addition of sodium azide,
levels of component C5a C5a itself exerts potent
in amounts comparable to its concentration as a preserva-
chemotactic and pro-inflammatory effects and its plasma
tive, to cultures of endothelial cells resulted in a decrease of
levels have been associated with increased cardiovascular
migration, proliferation and angiogenic properties of these
risk in patients with advanced atherosclerosis
cells In SMCs, addition of sodium azide (without
CRP is also involved in the inhibition of complement
CRP) did induce vasorelaxation and evoked inducible
activation through interaction with factor H (fH), which is
nitric oxide synthase (iNOS) induction and nitric monoxide
also present in injured areas. The CRP – fH complex
(NO) release. These effects were initially ascribed to the
interferes with the activity of C3b (see and
CRP. It is not clear yet how many of the reported effects of
thus will prevent formation of the MAC.
CRP can be ascribed to contamination and whether the
Through the interaction with complement factors, CRP
effects can be explained by contamination completely or
exerts a direct effect on arterial endothelial cells, by
whether there will simultaneous effects of contaminants and
increasing the expression of complement inhibitory factors
CRP. However, several studies showed direct effects of CRP
on the endothelial cells. This suggests that CRP-mediated
preparations that were free of contaminants, showing that
complement activation is a system set to regulate the
CRP may indeed have a direct role. Examples are studies
inflammatory reaction, because it will result in promoting
comparing the effect of plasma from CRP transgenic mice
the removal of debris from tissues and the deleterious effects
and wild-type mice or studies in which the effect of
of complement activation in patients with CVD
CRP containing a sodium azide contamination exceeds the
However, since complement activation also leads to the
effect of sodium azide alone which deliver strong
production of a variety of pro-inflammatory molecules, this
evidence of CRP having a direct and causal effect on several
mechanism of CRP-mediated complement regulation might
cell types and the inflammatory processes. Furthermore, the
also aggravate the inflammatory status in the entire body as
specific interaction of CRP with complement factors, cell
well as in the atherosclerotic plaque. Therefore, the direct
receptors, lipids and other inflammatory mediators assures
interaction between CRP and complement can both activate
the possibility of CRP being directly involved atheroscle-
and inhibit inflammation in atherosclerotic lesions.
E. Paffen, M.P.M. deMaat / Cardiovascular Research 71 (2006) 30 – 39
Fig. 1. Schematic representation of CRP-mediated complement regulation. Binding of CRP to microbial polysaccharides or ligands exposed on damagedactivates the classical pathway of complement. Activation is however limited to C1, C4, C2 and C3 with little consumption of C5 – C9. Surface-bound CRPrecruits fH, which regulates complement activation at the level of the alternate pathway C3 convertase and C5 convertase of both the classical and alternatepathways. fH is therefore thought to be responsible for low consumption of terminal pathway components during the CRP-initiated complement activation(adapted from Giannakis et al. fH: factor H, MBL: mannose-binding lectin pathway of complement, MAC: membrane attack complex.
5.2. Interaction with cell surface receptors
CRP through its interference with the binding of LDL toCD36
The close proximity of CRP to monocytic cells
in the arterial intima, attenuates its possibilities for a
direct contribution to the progression of atherosclerosis. The observation that CRP is localized between monocytes
Thrombosis contributes to the progression of the
underlines the possibility of a direct interaction of CRP
atherosclerotic lesion and to the precipitation of the
with these cells and with monocyte-derived macrophages
cardiovascular event. Direct actions of CRP which contrib-
via binding to a specific receptor. CRP binds to several
ute to the induction of a prothrombotic state may be the
receptors on human monocytes; to FcRgIIa (CD32) with
enhancement of the procoagulant activity or the
high affinity and to FcRgI (CD64) with lower affinity
reduction of fibrinolysis CRP has been suggested to
increasing phagocytosis and the release of inflam-
induce a prothrombotic state via induction of tissue factor
matory cytokines The Fc receptors have been
expression in human monocytes but only in the
described to mediate the effect of CRP on human aortic
presence of and through direct interaction with other blood
endothelial cells FcRgIIa is known as the putative
cells as T-lymphocytes, B-lymphocytes and natural killer
CRP receptor for leukocytes and also has been found
on bovine aortic endothelial cells CRP also binds to
In transgenic mice expressing human CRP (hCRP), the
the inhibitory receptor, FcgammaRIIb, blocking activating
injury-induced occlusion of the femoral artery (75% after 28
signals The binding of CRP to a receptor suggests
days) was enhanced compared to the amount of occlusion
its capacity to induce a specific biological effect
observed in wild-type mice (17% after 28 days)
such as direct involvement in cell-mediation and opsoni-
indicating a prothrombotic effect of hCRP. A direct effect
zation. Addition of CRP with and without anti-CD32-
of CRP on hemostasis was shown in a recent study, where
antibody to endothelial cells demonstrated the partial
recombinant human CRP was infused into human volun-
mediation of CRP in regulation of cell surface protein
teers, which resulted in the stimulation of both hemostasis
expression, such as the endothelial protein C receptor, by
CD32 However, the downstream effects of CRP
CRP may also inhibit fibrinolysis by increasing the
binding have not yet been elucidated. The interaction of
expression and activity of the main inhibitor of fibrinolysis,
CRP with CD36, a scavenger receptor which is expressed
plasminogen activator inhibitor-1 (PAI-1) in human aortic
by macrophages and is involved in uptake of low-density
endothelial cells (HAEC) Since PAI-1 promotes
lipoprotein particles (LDL), demonstrates a direct role of
atherothrombosis and progression of acute coronary syn-
E. Paffen, M.P.M. deMaat / Cardiovascular Research 71 (2006) 30 – 39
dromes, this effect of CRP may also affect CVD Also,
promotes MCP-1 mediated chemotaxis through upregula-
besides this effect on PAI-1, recently CRP has been
tion of CC chemokine receptor 2 expression in human
demonstrated to directly decrease antigen levels and the
activity of tissue plasminogen activator (tPa) in HAEC. tPa
The effect of CRP on T-lymphocytes is indirect. T-
is the substance normally inhibited by PAI-1. In this study,
lymphocytes are recruited to the atherosclerotic lesion as a
the direct and specific role of CRP is demonstrated by using
result of the ongoing inflammatory process. Through the
CRP that was free from sodium azide and LPS contamina-
stimulation of cytokine production and secretion by macro-
phages, CRP exerts an indirect effect on T-lymphocytespresent in the atherosclerotic lesion. CRP induces macro-
5.4. Cellular modulation, recruitment and activation
phages to express interleukin-12, which contributes to thedevelopment of CD4+ T-helper cells In turn, these cells
CRP contributes to an arterial pro-inflammatory and pro-
express interferon-g, which is synergistic with CRP in the
atherosclerotic phenotype by directly upregulating adhesion
execution of many functions contributing to the pro-
molecules and chemoattractant chemokines in endothelial
atherosclerotic phenotype. In contrast, during CRP induced
cells, vascular SMCs and monocytic cells. On the
activation of complement and opsonization of apoptotic
endothelial cell surface, expression of adhesion molecules
cells, the actively phagocyting macrophages reduce expres-
such as ICAM-1, VCAM-1, and E-selectin is upregulated
sion of IL-12 and thereby suppress T-lymphocytes
by CRP Via these processes, CRP induces plateletadhesion to endothelial cells CRP stimulates endo-
5.5. Expression of inflammatory mediators: cytokines,
thelial cell dysfunction and the recruitment of monocytes
and T-lymphocytes towards the endothelial wall. Thesefindings were reported by several groups, who also showed
CRP induces inflammatory cytokines in a dose-depen-
that CRP induced monocyte chemoattractant chemokine-1
dent way which provides further support for the
(MCP-1) production. This upregulation of adhesion mole-
hypothesis that interaction with mononuclear phagocytes
cules is partly mediated via the production of endothelin-1,
constitutes an important biological role for this acute phase
a potent endothelium-derived vasoactive factor, and by the
protein. Quantitative analysis of the CRP-induced release of
production of the inflammatory cytokines interleukin-6 and
interleukin-6, interleukin-1 and tumor necrosis factor-a by
interleukin-8. As to the effects of CRP on MCP-1
freshly isolated normal human monocytes, revealed slight
expression, aortic endothelial cells seem to be unresponsive
differences in time courses. All three cytokines were
whereas venous endothelial cells or monocytes
detected 4 h after CRP addition in vitro, with maximal
show increased expression of this chemoattractant. Since
levels of TNFa at 8 h and of interleukin-1 and interleukin-6
atherosclerosis mainly develops in the arteries, the clinical
significance of the effect of CRP on venous cells is not
Interleukin-8 (IL-8), a member of the CXC chemokines
promotes monocyte – endothelial cell adhesion and arrest
CRP is also known to activate the NF-nB signaling
and is abundant in atherosclerotic plaques. In human aortic
pathway in saphenous vein endothelial cells which, in
endothelial cells in vitro, CRP increases IL-8 protein and
recent light of possible azide contamination might be
mRNA expression in a time- and dose-dependent manner
an artefact. Also in vascular SMCs, CRP has been indicated
via specific upregulation of NF-nB activity
to activate NF-nB Therefore, CRP has been suggested
CRP induces production and secretion of MCP-1 in
to mediate proliferation and activation of vascular SMCs,
human umbilical vein endothelial cells but not in aortic
causing the accumulation of these cells in the vascular
endothelial cells MCP-1 present in the atherosclerotic
intima, which is a key event in the development of arterial
lesion can also originate from monocytes. CRP induces
lesions. Another manner in CRP directly affects the
a 7-fold increase in the production of monocyte MCP-1 in
activation and proliferation of vascular SMCs, is via
purified peripheral monocytes In patients with acute
upregulation of mRNA and protein and increased cell
coronary syndromes, baseline level of this chemoattractant
surface expression of the angiotensin type 1 receptor (AT1-
were elevated. This elevated expression is associated with
R). This was demonstrated in vitro in human vascular
both traditional risk factors for atherosclerosis as well as
SMCs and in vivo in a rat carotid artery angioplasty model
increased risk of myocardial infarction, independent of
CRP also appears to be involved in the infiltration of
In atherosclerotic lesions, CRP directly upregulates
monocytes into the vessel wall and their subsequent
mRNA expression of the macrophage markers CD11b and
development into foam cells. The deposition of CRP in
HLA-DR, as well as their protein products
the arterial wall precedes monocyte infiltration and direct
Monocyte expression of CD11b increased significantly
involvement of CRP in recruitment of blood monocytes
up to twofold when exposed to CRP, while no significant
has been demonstrated in vitro, suggesting CRP to be
difference in CD32 expression was observed, whereas CRP
chemotactic for human blood monocytes CRP also
exposure decreases CD31 expression. CRP can affect
E. Paffen, M.P.M. deMaat / Cardiovascular Research 71 (2006) 30 – 39
monocyte activation ex vivo and induce phenotypic changes
that result in an altered recruitment to endothelial cells
Another mechanism by which CRP influences the
CRP is directly involved in the process of apoptosis
development and maintenance of the athereosclerotic lesion
It binds to apoptotic cells in a Ca2+-dependent
is its involvement in the CD40 – CD40Ligand (CD40L or
manner and augments the classical pathway of complement
CD154) interaction. CD40L, a 33-kDa activation-induced T-
activation but protects the cells from assembling the
lymphocyte surface glycoprotein, binds to CD40, a phos-
terminal complement components (C5 – C9). Furthermore,
phorylated glycoprotein expressed on B-lymphocytes, vas-
CRP enhances opsonization and phagocytosis of apoptotic
cular endothelial cells, monocytes, macrophages and
cells by macrophages associated with the expression of the
fibroblasts. Like CRP, the amount of soluble CD40
anti-inflammatory cytokine transforming growth factor-h.
increases during inflammation and in the atherosclerotic
CRP and the classical complement components act in
lesion. Therefore CD40L has been suggested to be a marker
concert to promote non-inflammatory clearance of apoptotic
for inflammation and involved in risk of cardiovascular
cells The inhibitory effect of CRP on the NO
events as well CRP upregulates the cell surface
expression of endothelial progenitor cells directly inhibits
expression of CD40 and CD40L on human umbilical
their mobilization and differentiation, survival and function,
endothelial cells in time CD40L is shed into the
whereby it facilitates EC apoptosis and blocks the process of
vasculature. Elevated levels of this soluble CD40L
angiogenesis Apoptosis of vascular SMCs also plays
(sCD40L) identify patients with acute coronary syndromes
an important role in progression of atherosclerotic lesions
at increased risk of recurrent MI and death, independent of
and contributes to increased plaque vulnerability. Silencing
other variables as cardiac troponine T or CRP
the CRP-regulated GADD153 gene in vascular SMCsindicated that CRP plays an essential role in induced
CRP also binds to phosphatidylcholines, by which it
CRP has been described to decrease the expression and
participates directly in activation of macrophages and
bioactivity of endothelial nitric oxide synthase (eNOS or
neutrophils in the clearance of apoptotic and necrotic cells
NOS3) which results in reduced bioavailability of
However, neutrophils are not present in the
nitric monoxide (NO) and a subsequent effect of vasodila-
tation. It was demonstrated recently that this effect can becaused by sodium azide as well however, it is not clear
whether there remains a role for CRP. It is thereforeuncertain whether there is a causal role for CRP in the
The interaction between lipids and CRP is diverse. It has
regulation of expression of NO and involvement in vascular
been suggested that CRP could be the factor that links
reactivity Nevertheless, CRP has been suggested to
lipoprotein-deposition and complement activation in ath-
exert a specific effect on endothelial eNOS expression
erosclerotic plaques. Binding of tissue-deposited CRP to
through binding to the CRP receptor FcRgIIa In
enzymatically degraded LDL enhances complement activa-
HAECs and human coronary artery endothelial cells
tion, which may be relevant to the development and
(HCAECs), CRP contributes to a proatherogenic and
progression of the atherosclerotic lesion, particularly at
prothrombotic state by decreasing the release of NO and
early stages of atherosclerosis when low concentrations of
of the vasodilator and inhibitor of platelet aggregation
enzymatically degraded LDL are present And,
prostacyclin (PGI2), through directly increasing both
although direct involvement of CRP has not been demon-
strated, through this binding of CRP to enzymatically
In vascular smooth muscle cells, CRP reduces expression
degraded LDL, CRP may be involved in the massive
of the inducible variant of nitric oxide synthase (iNOS)
release of MCP-1 from macrophages described to be caused
and subsequent NO-synthesis as well. But again, this might
be an artefact caused by sodium azide In these vascular
Although the reports on interaction between CRP and ox-
SMCs, CRP also has been described to induce activation of
LDL are conflicting, complement activation as a result of
the iNOS promoter which, despite the fact that CRP
this interaction is generally considered unlikely. Neverthe-
seems to be no more than a weak inducer of NO-production,
less, CRP has been described to directly induce lectin-like
contradicts the study of Ikeda and coworkers. Nevertheless,
ox-LDL receptor-1 expression (LOX-1) in human aortic
both studies demonstrate that CRP is likely to be involved in
ECs, because this could be reduced with antibodies against
the regulation of cellular NO-levels. Furthermore, interac-
CD32/CD64, ET-1 or IL-6 Via LOX-1, CRP is
tion between CRP and interferon-gamma appears to
suggested to regulate monocytes adhesion to ECs and
enhance the effect of CRP on NO-regulation, which
indicates that there may be a direct effect of CRP because
The majority of sub-endothelial foam cells show
contaminants will not be able to exert these specific
positive staining for CRP. Zwaka et al. demonstrated that
native LDL that was co-incubated with CRP was taken up
E. Paffen, M.P.M. deMaat / Cardiovascular Research 71 (2006) 30 – 39
by macrophages via macropinocytosis. It was concluded
process was not possible but we discussed above that
that foam cell formation in human atherogenesis might be
one thing does not exclude the other.
caused in part by uptake of CRP-opsonized native LDL
Recently, several studies have reported conflicting results
on the direct contribution of CRP to atherosclerosis in mice
High levels of high-density lipoprotein (HDL) are
that transgenically expressed CRP. Since CRP is expressed
atheroprotective since HDL is involved in transporting
only at a very low concentration in mice and does not show
cholesterol from the periphery to the liver. HDL might also
an acute phase behavior, it is possible to study the role of
protect the endothelium since the CRP-induced upregulation
transgenic CRP in mice. The first report was from Paul and
of inflammatory adhesion molecules in HUVECs was
coworkers who observed larger aortic atherosclerotic lesions
completely blocked by HDL. So, HDL neutralizes CRP
in human CRP transgenic apolipoprotein (apo) E-knockout
induced proinflammatory activity HDL also inhibits
mice than in control mice but in this study the CRP
atherosclerosis through prevention of oxidation of LDL. It is
levels were very high. Recently, we did not see a difference
not known whether CRP has an effect on the oxidative
in lesion size or severity of atherosclerosis at the aortic root
between apoE*3-Leiden mice that did express human CRP(< 10 mg/L) and controls Reifenberg and coworkersreported similar observations in apoE-knockout mice
that expressed rabbit CRP and subsequently discussedwhether transgenic apoE-knockout mice would provide an
CRP may contribute to development of the atheroscle-
answer about the role of CRP in atherogenesis at all. In our
rotic lesion and the subsequent acute cardiovascular events
opinion, we believe that more studies are needed to
via its role in a large number of biological pathways. And
elucidate the differences, and we cannot ignore previous
although a number of the effects of CRP may be clouded by
studies in which mouse models were used to clearly
contamination, we demonstrated that a direct role of CRP in
demonstrate a direct role of CRP in atherogenesis
many inflammatory processes is probable. We reviewed the
In conclusion, it is now an established fact that elevated
roles of CRP in complement activation, cell adhesion and
CRP levels are associated with a worse prognosis for CVD,
recruitment, thrombosis, the expression of regulatory
such as myocardial infarction, stroke and unstable angina.
cytokines, apoptosis and lipids. All these mechanisms are
But it is important to distinguish between a role as a marker
part of or are compromised by the process of inflammation.
and a factor that directly causes a biological effect because
CRP may thus contribute to the development of the
this will determine the optimal therapeutic intervention.
atherosclerotic lesion via a direct pro-inflammatory effect.
CRP may be a causal factor as well as a marker for
CRP increases the release of endothelin-1 and upregulates
inflammation, depending on the concentration. This concen-
adhesion molecules and chemoattractant chemokines in
tration of CRP depends on the rates of production and
endothelial cells and vascular SMC. Most studies focus on
clearance. The fact that CRP is a very stable protein, which is
the effects of CRP on aortic endothelial cells but a few
not consumed to a significant extent in any process, and the
studies have also determined the effect on venous endothe-
clearance of which is not influenced by any known condition
lial cells as HUVECS and it was observed that CRP induced
is in agreement with its functioning as a causal factor,
the expression of MCP-1 CD40 and increased
attempting to prolong the stability of the atherosclerotic
activity of NF-nB This suggests that CRP exerts
different and specific effect on different cell types.
In the atherosclerotic lesion, many processes involving
However, it is important to realize that CRP also exerts
CRP and many different cell types have been described but
activity in anti-inflammatory or so-called protective mech-
there is also concern that part of the observed effects are not
anisms, such as suppressing the formation of the C5 – C9
mediated by CRP itself, but are the result of contamination of
complex. Thereby CRP is capable of maintaining a certain
the preparations with endotoxin or azide. Therefore, currently
balance in the inflammatory process and stability of the
the exact role of CRP in the initiation and progression of
atherosclerotic lesion. In the lesion, CRP, which stimulates
atherosclerosis is still unclear and needs to be further studied.
immunity as well as inflammation, contributes to theprolongation of the stability of the plaque.
A low level of chronic inflammation is represented by
CRP levels that are only slightly, but for a prolonged period,increased and these levels characterize CRP as a predictor of
We thank Prof. R. M. Bertina for critical reading of the
a cardiovascular event. On the other hand, a high
concentration of CRP (> 10 mg/L) can be measured for ashorter period during the acute phase where it is involved in
the inflammatory defense process. It has been suggestedthat, because of this expression of high levels of CRP, a
[1] Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med
direct biological or a causal function in the atherosclerotic
E. Paffen, M.P.M. deMaat / Cardiovascular Research 71 (2006) 30 – 39
[2] Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis.
[22] Kasper HU, Schmidt A, Roessner A. Expression of the adhesion
molecules ICAM, VCAM, and ELAM in the arteriosclerotic plaque.
[3] Gabay C, Kushner I. Acute-phase proteins and other systemic
Gen Diagn Pathol 1996;141:289 – 94.
responses to inflammation. N Engl J Med 1999;340:448 – 54.
[23] Ross R. Atherosclerosis is an inflammatory disease. Am Heart J
[4] Uhlar CM, Whitehead AS. Serum amyloid A, the major vertebrate
acute-phase reactant. Eur J Biochem 1999;265:501 – 23.
[24] Libby P, Sukhova G, Lee RT, Galis ZS. Cytokines regulate vascular
[5] Black S, Kushner I, Samols D. C-reactive protein. J Biol Chem
functions related to stability of the atherosclerotic plaque. J Cardio-
vasc Pharmacol 1995;25(Suppl 2):S9 – 12.
[6] Danesh J, Collins R, Appleby P, Peto R. Association of fibrinogen,
[25] Seifert PS, Hansson GK. Complement receptors and regulatory
C-reactive protein, albumin, or leukocyte count with coronary heart
proteins in human atherosclerotic lesions. Arteriosclerosis 1989;
disease: meta-analyses of prospective studies. JAMA 1998;
[26] Speidl WS, Exner M, Amighi J, Kastl SP, Zorn G, Maurer G, et al.
[7] Koenig W, Sund M, Frohlich M, Fischer HG, Lowel H, Doring A, et
Complement component C5a predicts future cardiovascular events in
al. C-reactive protein, a sensitive marker of inflammation, predicts
patients with advanced atherosclerosis. Eur Heart J 20052294 – 9.
future risk of coronary heart disease in initially healthy middle-aged
[27] Kassirer M, Zeltser D, Prochorov V, Schoenman G, Frimerman A,
men: results from the MONICA (Monitoring Trends and Determi-
Keren G, et al. Increased expression of the CD11b/CD18 antigen on
nants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to
the surface of peripheral white blood cells in patients with ischemic
1992. Circulation 1999;99:237 – 42.
heart disease: further evidence for smoldering inflammation in
[8] Ridker PM, Rifai N, Stampfer MJ, Hennekens CH. Plasma concen-
patients with atherosclerosis. Am Heart J 1999;138:555 – 9.
tration of interleukin-6 and the risk of future myocardial infarction
[28] Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of
among apparently healthy men. Circulation 2000;101:1767 – 72.
matrix metalloproteinases and matrix degrading activity in vulnerable
[9] Ridker PM, Buring JE, Shih J, Matias M, Hennekens CH.
regions of human atherosclerotic plaques. J Clin Invest 1994;
Prospective study of C-reactive protein and the risk of future
cardiovascular events among apparently healthy women. Circulation
[29] Calabro P, Willerson JT, Yeh ET. Inflammatory cytokines stimulated
C-reactive protein production by human coronary artery smooth
[10] Gill R, Kemp JA, Sabin C, Pepys MB. Human C-reactive protein
muscle cells. Circulation 2003;108:1930 – 2.
increases cerebral infarct size after middle cerebral artery occlusion
[30] Lombardo A, Biasucci LM, Lanza GA, Coli S, Silvestri P, Cianflone
in adult rats. J Cereb Blood Flow Metab 2004;24:1214 – 8.
D, et al. Inflammation as a possible link between coronary and
[11] Libby P, Ridker PM. Inflammation and atherosclerosis: role of C-
carotid plaque instability. Circulation 2004;109:3158 – 63.
reactive protein in risk assessment. Am J Med 2004;116(Suppl
[31] Torzewski J, Torzewski M, Bowyer DE, Frohlich M, Koenig W,
Waltenberger J, et al. C-reactive protein frequently colocalizes with
[12] Pepys MB, Hirschfield GM. C-reactive protein and atherothrombo-
the terminal complement complex in the intima of early atheroscle-
rotic lesions of human coronary arteries. Arterioscler Thromb Vasc
[13] Sesso HD, Buring JE, Rifai N, Blake GJ, Gaziano JM, Ridker PM.
C-reactive protein and the risk of developing hypertension. JAMA
[32] Zwaka TP, Hombach V, Torzewski J. C-reactive protein-mediated
low density lipoprotein uptake by macrophages: implications for
[14] Ridker PM, Rifai N, Rose L, Buring JE, Cook NR. Comparison of C-
atherosclerosis. Circulation 2001;103:1194 – 7.
reactive protein and low-density lipoprotein cholesterol levels in the
[33] Tillett WS, Francis T Jr. Serological reactions in pneumonia with a
nonprotein somatic fraction of pneumococcus. J Exp Med 1930;
[15] Doggen CJ, Berckmans RJ, Sturk A, Manger Cats V, Rosendaal FR.
[34] Castell JV, Gomez-Lechon MJ, David M, Fabra R, Trullenque R,
C-reactive protein, cardiovascular risk factors and the association
Heinrich PC. Acute-phase response of human hepatocytes: regulation
with myocardial infarction in men. J Intern Med 2000;248:406 – 14.
of acute-phase protein synthesis by interleukin-6. Hepatology
[16] Haverkate F, Thompson SG, Pyke SD, Gallimore JR, Pepys MB.
Production of C-reactive protein and risk of coronary events in stable
[35] Ishikawa T, Imamura T, Hatakeyama K, Date H, Nagoshi T,
and unstable angina. European Concerted Action on Thrombosis and
Kawamoto R, et al. Possible contribution of C-reactive protein
Disabilities Angina Pectoris Study Group. Lancet 1997;349:462 – 6.
within coronary plaque to increasing its own plasma levels across
[17] Ridker PM, Hennekens CH, Buring JE, Rifai N. C-reactive protein
coronary circulation. Am J Cardiol 2004;93:611 – 4.
and other markers of inflammation in the prediction of cardiovascular
[36] Kuta AE, Baum LL. C-reactive protein is produced by a small
disease in women. N Engl J Med 2000;342:836 – 43.
number of normal human peripheral blood lymphocytes. J Exp Med
[18] Liu C, Wang S, Deb A, Nath KA, Katusic ZS, McConnell JP, et al.
Proapoptotic, antimigratory, antiproliferative, and antiangiogenic
[37] Yasojima K, Schwab C, McGeer EG, McGeer PL. Generation of C-
effects of commercial C-reactive protein on various human endothe-
reactive protein and complement components in atherosclerotic
lial cell types in vitro: implications of contaminating presence of
plaques. Am J Pathol 2001;158:1039 – 51.
sodium azide in commercial preparation. Circ Res 2005;97:135 – 43.
[38] Shrive AK, Cheetham GM, Holden D, Myles DA, Turnell WG,
[19] Taylor KE, Giddings JC, van den Berg CW. C-reactive protein-
Volanakis JE, et al. Three dimensional structure of human C-reactive
induced in vitro endothelial cell activation is an artefact caused by
protein. Nat Struct Biol 1996;3:346 – 54.
azide and lipopolysaccharide. Arterioscler Thromb Vasc Biol 2005;
[39] Verma S, Szmitko PE, Yeh ET. C-reactive protein: structure affects
function. Circulation 2004;109:1914 – 7.
[20] Bisoendial RJ, Kastelein JJ, Levels JH, Zwaginga JJ, van den
[40] Diehl EE, Haines GK, Radosevich JA, Potempa LA. Immunohisto-
Boogaard B, Reitsma PH, et al. Activation of inflammation and
chemical localization of modified C-reactive protein antigen in
coagulation after infusion of C-reactive protein in humans. Circ Res
normal vascular tissue. Am J Med Sci 2000;319:79 – 83.
[41] Khreiss T, Jozsef L, Potempa LA, Filep JG. Conformational
[21] Singh U, Devaraj S, Jialal I. C-reactive protein decreases tissue
rearrangement in C-reactive protein is required for proinflammatory
plasminogen activator activity in human aortic endothelial cells:
actions on human endothelial cells. Circulation 2004;109:2016 – 22.
evidence that C-reactive protein is a procoagulant. Arterioscler
[42] Devaraj S, Venugopal S, Jialal I. Native pentameric C-reactive
Thromb Vasc Biol 2005;25:2216 – 21.
protein displays more potent pro-atherogenic activities in human
E. Paffen, M.P.M. deMaat / Cardiovascular Research 71 (2006) 30 – 39
aortic endothelial cells than modified C-reactive protein, Atheroscle-
[63] Bharadwaj D, Stein MP, Volzer M, Mold C, Du Clos TW. The major
rosis; in press, Corrected Proof, Available online 13 May 2005.
receptor for C-reactive protein on leukocytes is fcgamma receptor II.
[43] Schwedler SB, Amann K, Wernicke K, Krebs A, Nauck M, Wanner
C, et al. Native C-reactive protein increases whereas modified C-
[64] Escribano-Burgos M, Lopez-Farre A, del Mar GM, Macaya C,
reactive protein reduces atherosclerosis in apolipoprotein E-knockout
Garcia-Mendez A, Mateos-Caceres PJ, et al. Effect of C-reactive
mice. Circulation 2005;112:1016 – 23.
protein on Fcgamma receptor II in cultured bovine endothelial cells.
[44] Pepys MB, Hawkins PN, Kahan MC, Tennent GA, Gallimore JR,
Graham D, et al. Proinflammatory effects of bacterial recombinant
[65] Nan B, Yang H, Yan S, Lin PH, Lumsden AB, Yao Q, et al. C-
human C-reactive protein are caused by contamination with
reactive protein decreases expression of thrombomodulin and
bacterial products, not by C-reactive protein itself. Circ Res 2005;
endothelial protein C receptor in human endothelial cells. Surgery
[45] Osterud B. Interaction of endotoxins, blood elements and the vessel
[66] Penn MS, Topol EJ. Tissue factor, the emerging link between
wall. Prog Clin Biol Res 1985;189:67 – 79.
inflammation, thrombosis, and vascular remodeling. Circ Res 2001;
[46] Guha M, Mackman N. LPS induction of gene expression in human
monocytes. Cell Signal 2001;13:85 – 94.
[67] Libby P, Simon DI. Inflammation and thrombosis: the clot thickens.
[47] Rivers RP, Hathaway WE, Weston WL. The endotoxin-induced
coagulant activity of human monocytes. Br J Haematol 1975;
[68] Juhan-Vague I, Pyke SD, Alessi MC, Jespersen J, Haverkate F,
Thompson SG. Fibrinolytic factors and the risk of myocardial
[48] Jungi TW, Miserez R, Brcic M, Pfister H. Change in sensitivity to
infarction or sudden death in patients with angina pectoris. ECAT
lipopolysaccharide during the differentiation of human monocytes to
Study Group. European Concerted Action on Thrombosis and
macrophages in vitro. Experientia 1994;50:110 – 4.
Disabilities. Circulation 1996;94:2057 – 63.
[49] Mackman N. Lipopolysaccharide induction of gene expression in
[69] Cushman M, Lemaitre RN, Kuller LH, Psaty BM, Macy EM,
human monocytic cells. Immunol Res 2000;21:247 – 51.
Sharrett AR, et al. Fibrinolytic activation markers predict myocardial
[50] Nagoshi Y, Kuwasako K, Cao YN, Kitamura K, Eto T. Effects of C-
infarction in the elderly. The Cardiovascular Health Study. Arte-
reactive protein on atherogenic mediators and adrenomedullin in
rioscler Thromb Vasc Biol 1999;19:493 – 8.
human coronary artery endothelial and smooth muscle cells.
[70] Whisler RL, Proctor VK, Downs EC, Mortensen RF. Modulation of
Biochem Biophys Res Commun 2004;314:1057 – 63.
human monocyte chemotaxis and procoagulant activity by human C-
[51] van den Berg CW, Taylor KE, Lang D. C-reactive protein-induced in
reactive protein (CRP). Lymphokine Res 1986;5:223 – 8.
vitro vasorelaxation is an artefact caused by the presence of sodium
[71] Nakagomi A, Freedman SB, Geczy CL. Interferon-gamma and
azide in commercial preparations. Arterioscler Thromb Vasc Biol
lipopolysaccharide potentiate monocyte tissue factor induction by
C-reactive protein: relationship with age, sex, and hormone replace-
[52] Brill A, Yaron G, Dashevsky O, Danenberg H, Varon D. CRP
ment treatment. Circulation 2000;101:1785 – 91.
induces platelet adhesion to endothelial cells under flowPoster ISTH
[72] Paffen E, Vos HL, Bertina RM. C-reactive protein does not directly
induce tissue factor in human monocytes. Arterioscler Thromb Vasc
[53] Pepys MB, Hirschfield GM. C-reactive protein: a critical update.
[73] Danenberg HD, Szalai AJ, Swaminathan RV, Peng L, Chen Z, Seifert
[54] Volanakis JE, Kaplan MH. Interaction of C-reactive protein com-
P, et al. Increased thrombosis after arterial injury in human C-reactive
plexes with the complement system: II. Consumption of guinea pig
protein-transgenic mice. Circulation 2003;108:512 – 5.
complement by CRP complexes: requirement for human C1q.
[74] Lip GY, Blann AD, Farooqi IS, Zarifis J, Sagar G, Beevers DG.
Sequential alterations in haemorheology, endothelial dysfunction,
[55] Szalai AJ, van Ginkel FW, Wang Y, McGhee JR, Volanakis JE.
platelet activation and thrombogenesis in relation to prognosis
Complement-dependent acute-phase expression of C-reactive protein
following acute stroke: the West Birmingham Stroke Project. Blood
and serum amyloid P-component. J Immunol 2000;165:1030 – 5.
Coagul Fibrinolysis 2002;13:339 – 47.
[56] Giannakis E, Male DA, Ormsby RJ, Mold C, Jokiranta TS,
[75] Devaraj S, Xu DY, Jialal I. C-reactive protein increases plasminogen
Ranganathan S, et al. Multiple ligand binding sites on domain
activator inhibitor-1 expression and activity in human aortic
seven of human complement factor H. Int Immunopharmacol 2001;
endothelial cells: implications for the metabolic syndrome and
atherothrombosis. Circulation 2003;107:398 – 404.
[57] Szalai AJ, Agrawal A, Greenhough TJ, Volanakis JE. C-reactive
[76] Pasceri V, Wu HD, Willerson JT, Yeh ET. Modulation of vascular
protein: structural biology and host defense function. Clin Chem Lab
inflammation in vitro and in vivo by peroxisome proliferator-
activated receptor-gamma activators. Circulation 2000;101:235 – 8.
[58] Reynolds GD, Vance RP. C-reactive protein immunohistochemical
[77] Pasceri V, Chang J, Willerson JT, Yeh ET. Modulation of C-reactive
localization in normal and atherosclerotic human aortas. Arch Pathol
protein-mediated monocyte chemoattractant protein-1 induction in
human endothelial cells by anti-atherosclerosis drugs. Circulation
[59] Torzewski M, Rist C, Mortensen RF, Zwaka TP, Bienek M,
Waltenberger J, et al. C-reactive protein in the arterial intima: role
[78] Thomassen MJ, Meeker DP, Deodhar SD, Wiedemann HP, Barna BP.
of C-reactive protein receptor-dependent monocyte recruitment in
Activation of human monocytes and alveolar macrophages by a
atherogenesis. Arterioscler Thromb Vasc Biol 2000;20:2094 – 9.
synthetic peptide of C-reactive protein. J Immunother 1993;13:1 – 6.
[60] Crowell RE, Du Clos TW, Montoya G, Heaphy E, Mold C. C-
[79] Verma S, Badiwala MV, Weisel RD, Li SH, Wang CH, Fedak PW, et
reactive protein receptors on the human monocytic cell line U-937.
al. C-reactive protein activates the nuclear factor-kappaB signal
Evidence for additional binding to Fc gamma RI. J Immunol 1991;
transduction pathway in saphenous vein endothelial cells: implica-
tions for atherosclerosis and restenosis. J Thorac Cardiovasc Surg
[61] Marnell L, Mold C, Du Clos TW. C-reactive protein: ligands,
receptors and role in inflammation. Clin Immunol 2005;117:104 – 11.
[80] Hattori Y, Matsumura M, Kasai K. Vascular smooth muscle cell
[62] Devaraj S, Du Clos TW, Jialal I. Binding and internalization of C-
activation by C-reactive protein. Cardiovasc Res 2003;58:186 – 95.
reactive protein by Fcgamma receptors on human aortic endothelial
[81] Wang CH, Li SH, Weisel RD, Fedak PW, Dumont AS, Szmitko P, et
cells mediates biological effects. Arterioscler Thromb Vasc Biol
al. C-reactive protein upregulates angiotensin type 1 receptors in
vascular smooth muscle. Circulation 2003;107:1783 – 90.
E. Paffen, M.P.M. deMaat / Cardiovascular Research 71 (2006) 30 – 39
[82] Han KH, Hong KH, Park JH, Ko J, Kang DH, Choi KJ, et al. C-
[98] Ikeda U, Takahashi M, Shimada K. C-reactive protein directly
reactive protein promotes monocyte chemoattractant protein-1-
inhibits nitric oxide production by cytokine-stimulated vascular
mediated chemotaxis through upregulating CC chemokine receptor
smooth muscle cells. J Cardiovasc Pharmacol 2003;42:607 – 11.
2 expression in human monocytes. Circulation 2004;109:2566 – 71.
[99] Lafuente N, Azcutia V, Matesanz N, Cercas E, Rodriguez-Manas L,
[83] Yamashita H, Shimada K, Seki E, Mokuno H, Daida H. Concen-
Sanchez-Ferrer CF, et al. Evidence for sodium azide as an artifact
trations of interleukins, interferon, and C-reactive protein in stable
mediating the modulation of inducible nitric oxide synthase by C-
and unstable angina pectoris. Am J Cardiol 2003;91:133 – 6.
reactive protein. J Cardiovasc Pharmacol 2005;45:193 – 6.
[84] Kim SJ, Gershov D, Ma X, Brot N, Elkon KB. Opsonization of
[100] Blaschke F, Bruemmer D, Yin F, Takata Y, Wang W, Fishbein MC, et
apoptotic cells and its effect on macrophage and T cell immune
al. C-reactive protein induces apoptosis in human coronary vascular
responses. Ann NY Acad Sci 2003;987:68 – 78.
smooth muscle cells. Circulation 2004;110:579 – 87.
[85] Devaraj S, O’keefe G, Jialal I. Defining the pro-inflammatory
[101] Gershov D, Kim S, Brot N, Elkon KB. C-reactive protein binds to
phenotype using high sensitive C-reactive protein levels as the
apoptotic cells, protects the cells from assembly of the terminal
biomarker. J Clin Endocrinol Metab 2005;90:4549 – 54.
complement components, and sustains an antiinflammatory innate
[86] Ballou SP, Lozanski G. Induction of inflammatory cytokine release
immune response: implications for systemic autoimmunity. J Exp
from cultured human monocytes by C-reactive protein. Cytokine
[102] Du Clos TW. Function of C-reactive protein. Ann Med 2000;
[87] Devaraj S, Kumaresan PR, Jialal I. Effect of C-reactive protein on
chemokine expression in human aortic endothelial cells. J Mol Cell
[103] Chang MK, Binder CJ, Torzewski M, Witztum JL. C-reactive protein
binds to both oxidized LDL and apoptotic cells through recognition
[88] Nelken NA, Coughlin SR, Gordon D, Wilcox JN. Monocyte
of a common ligand: phosphorylcholine of oxidized phospholipids.
chemoattractant protein-1 in human atheromatous plaques. J Clin
Proc Natl Acad Sci U S A 2002;99:13043 – 8.
[104] Klouche M, Gottschling S, Gerl V, Hell W, Husmann M, Dorweiler
[89] de Lemos JA, Morrow DA. Combining natriuretic peptides and
B, et al. Atherogenic properties of enzymatically degraded LDL:
necrosis markers in the assessment of acute coronary syndromes. Rev
selective induction of MCP-1 and cytotoxic effects on human
Cardiovasc Med 2003;4(Suppl 4):S37 – 46.
macrophages. Arterioscler Thromb Vasc Biol 1998;18:1376 – 85.
[90] Woollard KJ, Philllips DC, Griffiths HR. Direct modulatory effect of
[105] Li L, Roumeliotis N, Sawamura T, Renier G. C-reactive protein
C-reactive protein on primary human monocyte adhesion to human
enhances LOX-1 expression in human aortic endothelial cells:
endothelial cells. Clin Exp Immunol 2002;130:256 – 62.
relevance of LOX-1 to C-reactive protein-induced endothelial
[91] Schonbeck U, Libby P. The CD40/CD154 receptor/ligand dyad. Cell
dysfunction. Circ Res 2004;95:877 – 83.
[106] Wadham C, Albanese N, Roberts J, Wang L, Bagley CJ, Gamble JR,
[92] Lin R, Liu J, Gan W, Yang G. C-reactive protein-induced expression
et al. High-density lipoproteins neutralize C-reactive protein proin-
of CD40 – CD40L and the effect of lovastatin and fenofibrate on it
flammatory activity. Circulation 2004;109:2116 – 22.
in human vascular endothelial cells. Biol Pharm Bull 2004;27:
[107] Pepys MB. CRP or not CRP? That is the question. Arterioscler
Thromb Vasc Biol 2005;25:1091 – 4.
[93] Varo N, Vicent D, Libby P, Nuzzo R, Calle-Pascual AL, Bernal MR,
[108] Paul A, Ko KW, Li L, Yechoor V, McCrory MA, Szalai AJ, et al. C-
et al. Elevated plasma levels of the atherogenic mediator soluble
reactive protein accelerates the progression of atherosclerosis in
CD40 ligand in diabetic patients: a novel target of thiazolidinediones.
apolipoprotein E-deficient mice. Circulation 2004;109:647 – 55.
[109] Trion A, de Maat MP, Jukema JW, van der Laarse A, Maas MC,
[94] Venugopal SK, Devaraj S, Yuhanna I, Shaul P, Jialal I. Demon-
Offerman EH, et al. No effect of C-reactive protein on early
stration that C-reactive protein decreases eNOS expression and
atherosclerosis development in apolipoprotein E*3-Leiden/human
bioactivity in human aortic endothelial cells. Circulation 2002;106:
C-reactive protein transgenic mice. Arterioscler Thromb Vasc Biol
[95] Swafford Jr AN, Bratz IN, Knudson JD, Rogers PA, Timmerman JM,
[110] Reifenberg K, Lehr HA, Baskal D, Wiese E, Schaefer SC, Black S, et
Tune JD, et al. C-reactive protein does not relax vascular smooth
al. Role of C-reactive protein in atherogenesis: can the apolipoprotein
muscle: effects mediated by sodium azide in commercially available
E knockout mouse provide the answer? Arterioscler Thromb Vasc
preparations. Am J Physiol, Heart Circ Physiol 2005;288:H1786 – 95.
[96] Clapp BR, Hirschfield GM, Storry C, Gallimore JR, Stidwill RP,
[111] Ursella S, Mazzone M, Portale G, Testa A, Pignataro G, Covino M, et
Singer M, et al. Inflammation and endothelial function: direct
al. How to use the C-reactive protein in cardiac disease? Minerva
vascular effects of human C-reactive protein on nitric oxide
bioavailability. Circulation 2005;111:1530 – 6.
[97] Venugopal SK, Devaraj S, Jialal I. C-reactive protein decreases
prostacyclin release from human aortic endothelial cells. Circulation2003;108:1676 – 8.
CENTRE DE REFERENCE DES MALADIES OSSEUSES CONSTITUTIONNELLES PRISE EN CHARGE D’UN PATIENT ATTEINT D’UNE FIBRODYSPLASIE OSSIFIANTE PROGRESSIVE La Fibrodysplasie Ossifiante Progressive (FOP) se caractérise par une ossification progressive des muscles et des tendons, le plus souvent précédée de poussées inflammatoires, selon une progression crânio-caudale. Elle est toujours as
8945d_043-044 6/18/03 11:21 AM Page 43 mac85 Mac 85:1st shift: 1268_tm:8945d: SENDING A SIGNAL THROUGH A GAS For decades scientists have tried to understand how cells work together in tis-sues, as well as in whole organisms. By the 1980s, the identity of many signal-ing molecules, the cellular responses they evoked, and many aspects of intracellu-lar signaling pathways were understood. All t

 Cardiovascular Research 71 (2006) 30 – 39
C-reactive protein in atherosclerosis: A causal factor?
aHemostasis and Thrombosis Research Centre, Dept. of Hematology, Leiden University Medical Centre, Leiden, The Netherlands
bDepartment of Hematology, Room Ee 13.93, Erasmus University Medical Center, Dr. Molewaterplein 50, 3015 GE Rotterdam, The Netherlands
Received 3 August 2005; received in revised form 3 March 2006; accepted 6 March 2006
Atherosclerosis is considered a to be multifactorial disease driven by inflammatory reactions. The process of inflammation also
contributes to the pathogenesis of acute atherothrombotic events. C-reactive protein (CRP) is an acute phase protein and its concentration inserum reflects the inflammatory condition of the patient. Levels of CRP are consistently associated with cardiovascular disease (CVD) andpredict myocardial infarctions and stroke. Since CRP is present in the atherosclerotic lesion, it may actively contribute to the progression and/or instability of the atherosclerotic plaque. The role of CRP in inflammation and its causality in atherosclerosis are the subject of manyinvestigations but are not yet fully elucidated. This review focuses on recently identified mechanisms by which CRP may modulate andevolve the process of atherosclerosis. We discuss the function of CRP and review the most recent evidence for an independent role of CRP inthe development of atherosclerosis. Many studies suggest such a role, but a number of the described effects may be the result ofcontamination of the CRP preparations.
Cardiovascular Research 71 (2006) 30 – 39
C-reactive protein in atherosclerosis: A causal factor?
aHemostasis and Thrombosis Research Centre, Dept. of Hematology, Leiden University Medical Centre, Leiden, The Netherlands
bDepartment of Hematology, Room Ee 13.93, Erasmus University Medical Center, Dr. Molewaterplein 50, 3015 GE Rotterdam, The Netherlands
Received 3 August 2005; received in revised form 3 March 2006; accepted 6 March 2006
Atherosclerosis is considered a to be multifactorial disease driven by inflammatory reactions. The process of inflammation also
contributes to the pathogenesis of acute atherothrombotic events. C-reactive protein (CRP) is an acute phase protein and its concentration inserum reflects the inflammatory condition of the patient. Levels of CRP are consistently associated with cardiovascular disease (CVD) andpredict myocardial infarctions and stroke. Since CRP is present in the atherosclerotic lesion, it may actively contribute to the progression and/or instability of the atherosclerotic plaque. The role of CRP in inflammation and its causality in atherosclerosis are the subject of manyinvestigations but are not yet fully elucidated. This review focuses on recently identified mechanisms by which CRP may modulate andevolve the process of atherosclerosis. We discuss the function of CRP and review the most recent evidence for an independent role of CRP inthe development of atherosclerosis. Many studies suggest such a role, but a number of the described effects may be the result ofcontamination of the CRP preparations.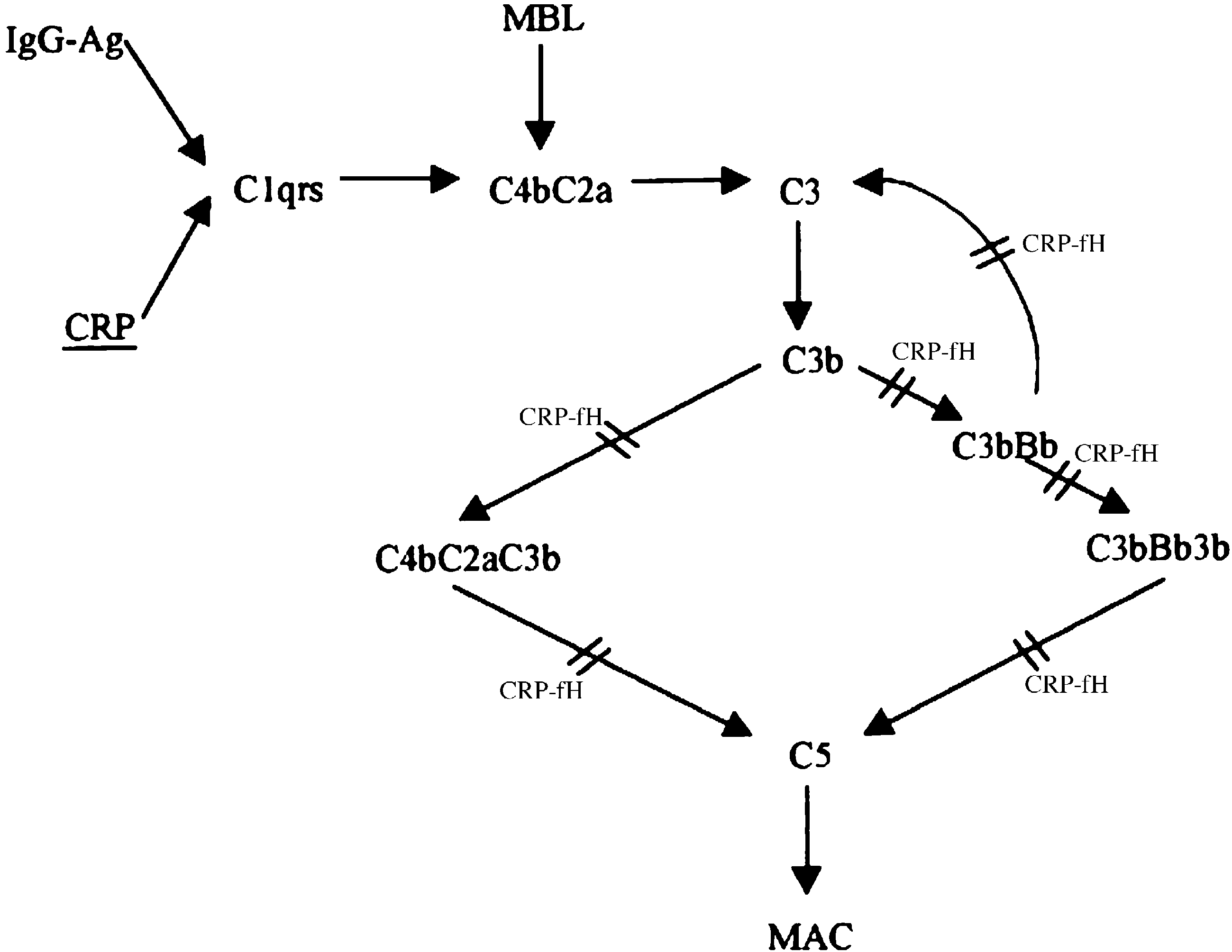 E. Paffen, M.P.M. deMaat / Cardiovascular Research 71 (2006) 30 – 39
Fig. 1. Schematic representation of CRP-mediated complement regulation. Binding of CRP to microbial polysaccharides or ligands exposed on damagedactivates the classical pathway of complement. Activation is however limited to C1, C4, C2 and C3 with little consumption of C5 – C9. Surface-bound CRPrecruits fH, which regulates complement activation at the level of the alternate pathway C3 convertase and C5 convertase of both the classical and alternatepathways. fH is therefore thought to be responsible for low consumption of terminal pathway components during the CRP-initiated complement activation(adapted from Giannakis et al. fH: factor H, MBL: mannose-binding lectin pathway of complement, MAC: membrane attack complex.
E. Paffen, M.P.M. deMaat / Cardiovascular Research 71 (2006) 30 – 39
Fig. 1. Schematic representation of CRP-mediated complement regulation. Binding of CRP to microbial polysaccharides or ligands exposed on damagedactivates the classical pathway of complement. Activation is however limited to C1, C4, C2 and C3 with little consumption of C5 – C9. Surface-bound CRPrecruits fH, which regulates complement activation at the level of the alternate pathway C3 convertase and C5 convertase of both the classical and alternatepathways. fH is therefore thought to be responsible for low consumption of terminal pathway components during the CRP-initiated complement activation(adapted from Giannakis et al. fH: factor H, MBL: mannose-binding lectin pathway of complement, MAC: membrane attack complex.